
SOGROYA 10 mg-1,5 ml, solution injectable en stylo prérempli, boîte de 1 stylo prérempli multidose de 1,50 ml
Dernière révision : 23/06/2025
Taux de TVA : 2.1%
Prix de vente : 402,97 €
Taux remboursement SS : 100%
Base remboursement SS : 402,97 €
Laboratoire exploitant : NOVO NORDISK
Source :

Sogroya est indiqué pour le traitement substitutif de l'hormone de croissance (GH*) endogène chez les enfants âgés de 3 ans et plus, et les adolescents présentant un retard de croissance dû à un déficit en hormone de croissance (GHD** pédiatrique), et chez les adultes présentant un déficit en hormone de croissance (GHD adulte).
*GH : growth hormone
**GHD : growth hormone deficiency
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Somapacitan ne doit pas être utilisé en cas de preuve d'une activité tumorale. Les tumeurs intracrâniennes doivent être inactives et tout traitement antitumoral devra être terminé avant de commencer le traitement par somapacitan. Le traitement doit être interrompu en cas de signes de croissance de la tumeur, voir rubrique Mises en garde spéciales et précautions d'emploi.
Somapacitan ne doit pas être utilisé pour favoriser la croissance longitudinale chez les enfants dont les épiphyses sont soudées, voir rubrique Posologie et mode d'administration.
Les patients en état critique aigu souffrant de complications après une intervention chirurgicale à cœur ouvert, une intervention chirurgicale de l'abdomen, un polytraumatisme accidentel, une insuffisance respiratoire aiguë ou des pathologies similaires ne doivent pas être traités par somapacitan (concernant les patients sous traitement de substitution, voir rubrique Mises en garde spéciales et précautions d'emploi).
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Insuffisance corticosurrénalienne
L'introduction du traitement par hormone de croissance peut entraîner une inhibition de la 11βHSD-1 et une diminution des concentrations de cortisol sérique. Chez les patients traités par hormone de croissance, un hypoadrénalisme central non préalablement diagnostiqué (secondaire) peut être révélé et un traitement substitutif par glucocorticoïdes peut être nécessaire. De plus, les patients sous traitement substitutif par glucocorticoïdes pour un hypoadrénalisme préalablement diagnostiqué peuvent nécessiter une augmentation de leurs doses d'entretien ou de leurs doses en cas de stress après l'initiation du traitement par hormone de croissance. Il est nécessaire de surveiller les patients présentant un hypoadrénalisme connu afin de détecter une réduction des taux de cortisol sérique et/ou la nécessité d'augmenter les doses de glucocorticoïdes, voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions.
Trouble du métabolisme du glucose
Le traitement par hormone de croissance peut diminuer la sensibilité à l'insuline, en particulier aux doses plus élevées chez les patients sensibles, et en conséquence, une hyperglycémie peut survenir chez les sujets ayant une capacité sécrétoire d'insuline inadéquate. Le traitement par hormone de croissance peut ainsi révéler la présence, jusque-là inconnue, d'une intolérance au glucose et d'un diabète sucré. En conséquence, les taux de glucose doivent être surveillés périodiquement chez tous les patients traités par hormone de croissance, en particulier chez ceux qui ont des facteurs de risque de diabète, tels que l'obésité, ou des antécédents familiaux de diabète. Les patients ayant un diabète de type 1 ou de type 2 pré-existant ou une intolérance au glucose doivent être surveillés étroitement pendant le traitement par hormone de croissance. Les doses de médicaments hypoglycémiants peuvent nécessiter un ajustement lorsqu'un traitement par hormone de croissance est instauré chez ces patients.
Tumeurs
Il n'y a pas de preuve d'un risque accru de nouveaux cancers primitifs chez les patients traités par hormone de croissance.
Chez les patients en rémission complète de pathologies malignes ou qui ont été traités pour des tumeurs bénignes, le traitement par hormone de croissance n'a pas été associé à un accroissement du taux de rechute.
Les patients ayant obtenu une rémission complète de pathologies malignes ou ayant été traités pour des tumeurs bénignes doivent être étroitement surveillés afin de détecter toute rechute après le début du traitement par hormone de croissance. Le traitement par hormone de croissance doit être interrompu en cas de développement ou de réapparition d'une tumeur bénigne ou maligne. Une augmentation globalement légère des néoplasmes secondaires a été observée chez les survivants de cancers pédiatriques traités par hormone de croissance, les plus fréquents étant les tumeurs intracrâniennes. Le principal facteur de risque de néoplasmes secondaires semble être l'exposition antérieure aux radiations.
Hypertension intracrânienne bénigne
En cas de céphalées sévères ou récurrentes, de symptômes visuels, de nausées et/ou vomissements, un examen de fond d'œil afin de dépister un éventuel œdème papillaire est recommandé. Si un œdème papillaire est confirmé, un diagnostic d'hypertension intracrânienne bénigne doit être envisagé et, le cas échéant, le traitement par hormone de croissance doit être interrompu. Il n'existe pas actuellement de preuves suffisantes pour guider la prise de décision clinique chez les patients ayant une hypertension intracrânienne résolue. Si le traitement par hormone de croissance est réinstauré, une surveillance étroite des symptômes d'hypertension intracrânienne est nécessaire.
Fonction thyroïdienne
L'hormone de croissance augmente la conversion extra-thyroïdienne de T4 en T3 et peut ainsi révéler une hypothyroïdie débutante. Étant donné que l'hypothyroïdie interfère avec la réponse au traitement par hormone de croissance, la fonction thyroïdienne des patients doit être régulièrement contrôlée et ils doivent recevoir un traitement de substitution par hormones thyroïdiennes si cela s'avère nécessaire, voir rubriques Interactions avec d'autres médicaments et autres formes d'interactions et Effets indésirables.
Utilisation avec les œstrogènes par voie orale
L'œstrogène par voie orale influence la réponse de l'IGF-1 à l'hormone de croissance, dont somapacitan.
Les patientes qui prennent toute forme d'œstrogène par voie orale (hormonothérapie ou contraception) doivent envisager de changer la voie d'administration de l'œstrogène (p. ex., hormonothérapie transdermique, vaginale) ou d'utiliser une autre forme de contraception. Si une femme sous œstrogène par voie orale débute un traitement par somapacitan, des doses initiales plus élevées et une période de titration plus longue peuvent être nécessaires (voir rubrique Posologie et mode d'administration).
Si une patiente traitée par somapacitan commence un traitement œstrogénique par voie orale, il peut être nécessaire d'augmenter la dose de somapacitan pour maintenir les taux sériques d'IGF-1 dans l'intervalle normal pour l'âge. Inversement, si une patiente sous somapacitan arrête son traitement œstrogénique par voie orale, il est possible que la dose de somapacitan doive être réduite pour éviter un excès de somapacitan et/ou des effets indésirables, voir rubriques Posologie et mode d'administration et Interactions avec d'autres médicaments et autres formes d'interactions.
Affections de la peau et du tissu sous-cutané
Lorsque somapacitan est administré au même endroit durant une période prolongée, des modifications locales du tissu sous-cutané telles qu'une lipohypertrophie, une lipoatrophie et une lipodystrophie acquise peuvent survenir. Le site d'injection doit être changé à chaque injection pour minimiser ce risque, voir rubriques Posologie et mode d'administration et Effets indésirables.
Anticorps
Il n'a pas été observé d'anticorps anti-somapacitan chez les patients adultes présentant un GHD. Quelques patients pédiatriques présentant un GHD ont été testés positifs à des anticorps liants somapacitan. Aucun de ces anticorps n'était neutralisant et aucun impact sur les effets cliniques n'a été observé. Une recherche d'anticorps anti-somapacitan doit être réalisée chez les patients qui ne répondent pas au traitement.
État critique aigu
Les effets de l'hormone de croissance sur la récupération ont été étudiés dans deux essais contrôlés versus placebo chez 522 patients adultes en état critique à la suite de complications secondaires à une intervention chirurgicale à cœur ouvert, une intervention chirurgicale abdominale, un polytraumatisme accidentel ou une insuffisance respiratoire aiguë. La mortalité était plus élevée chez les patients traités par 5,3 ou 8 mg d'hormone de croissance par jour par rapport aux patients recevant un placebo, 42 % vs 19 %. Sur la base de ces informations, ces types de patients ne doivent pas être traités par somapacitan. En l'absence d'informations sur la sécurité du traitement substitutif par hormone de croissance chez les patients en état critique aigu, le bénéfice de la poursuite du traitement dans cette situation doit être évalué par rapport aux risques potentiels associés.
Le déficit en hormone de croissance chez l'adulte est une maladie chronique qui doit être traitée en conséquence ; toutefois, l'expérience chez les patients de plus de 60 ans et chez les patients traités pendant plus de cinq ans pour un déficit en hormone de croissance sont encore limitées.
Pancréatite
Quelques cas de pancréatite ont été rapportés au cours d'un traitement par d'autres médicaments à base d'hormone de croissance. Cela doit être envisagé chez les patients traités par somapacitan qui développent des douleurs abdominales inexpliquées.
Epiphysiolyse de la tête fémorale
Chez les enfants avec une croissance rapide et les patients avec des
troubles endocriniens, incluant le GHD, l'épiphysiolyse de la tête
fémorale peut survenir plus fréquemment que dans la population
générale. Les enfants avec des douleurs persistantes à la hanche/au
genou et/ou présentant un boitement pendant le traitement avec du
somapacitan doivent être examinés cliniquement.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c'est-à-dire essentiellement « sans sodium ».
Résumé du profil de sécurité
Les effets indésirables (EI) les plus fréquemment rapportés sont (par ordre décroissant de fréquence [GHD pédiatrique, GHD adulte]) les céphalées (12 %, 12 %), les douleurs dans les extrémités (9 %, NA), l'hypothyroïdie (5 %, 2 %), les réactions au site d'injection (5 %, 1 %), l'œdème périphérique (3 %, 4 %), l'arthralgie (2 %, 7 %), l'hyperglycémie (2 %, 1 %), la fatigue (2 %, 6 %) et l'insuffisance corticosurrénalienne (1,5 %, 3 %).
Tableau des effets indésirables
Les EI mentionnés dans le tableau 2 sont basés sur les données de sécurité issues d'un essai pivot de phase 3 en cours (52 semaines) chez des patients pédiatriques présentant un GHD (âge lors de l'initiation : 2,5 à 11 ans) ayant développé des effets indésirables après traitement par somapacitan. Les fréquences des EI ont été calculées sur la base des fréquences observées dans l'essai pivot de phase 3.
Les effets indésirables mentionnés dans le tableau 3 sont basés sur les données de sécurité compilées de trois essais de phase 3 conduits chez des patients adultes présentant un GHD (âge lors de l'initiation : 19 à 77 ans).
Les EI sont listés par classe de systèmes d'organes MedDRA et catégorie de fréquence définie comme : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000).
Tableau 2 : Effets indésirables basés sur l'essai clinique de phase 3 chez les patients pédiatriques présentant un GHD
|
Classe de systèmes d'organes
MedDRA |
Très fréquent | Fréquent |
| Affections endocriniennes |
|
Hypothyroïdie* Insuffisance corticosurrénalienne |
|
Troubles du métabolisme et de la nutrition |
|
Hyperglycémie |
| Affections du système nerveux | Céphalées* |
|
|
Affections musculo squelettiques et systémiques |
|
Arthralgie Douleur dans les extrémités** |
|
Troubles généraux et anomalies au site d'administration |
|
Œdème périphérique* Réactions au site d'injection*# Fatigue |
#Les réactions au site d'injection incluaient une ecchymose au site d'injection (1,5 %), une douleur au site d'injection (1,5 %), un hématome au site d'injection (1,5 %) et un gonflement au site d'injection (0,8 %).
** Principalement une douleur légère à la jambe
Tableau 3 : Effets indésirables basés sur trois essais de phase 3 terminés chez des patients
adultes présentant un GHD
|
Classe de systèmes d'organes MedDRA |
Très fréquent |
Fréquent |
Peu fréquent |
|
Affections endocriniennes |
|
Insuffisance corticosurrénalienne
Hypothyroïdie |
|
|
Troubles
du métabolisme et de la nutrition |
|
Hyperglycémie* |
|
|
Affections
du système nerveux |
Céphalées |
Paresthésie |
Syndrome
du canal carpien |
|
Affections
de la peau et du tissu sous-cutané |
|
Rash* Urticaire* |
Lipohypertrophie* Prurit* |
|
Affections musculo- squelettiques
et systémiques |
|
Arthralgie Myalgie Raideur musculaire* |
Raideur articulaire |
|
Troubles généraux et anomalies au site |
|
Œdème périphérique
Fatigue Asthénie Réactions au site d'injection* |
|
Description de certains effets indésirables
Œdème périphérique
L'œdème périphérique a été fréquemment observé (3 % dans le
GHD pédiatrique et 4 % dans le GHD adulte). Les patients présentant un déficit
en hormone de croissance sont caractérisés par un déficit du volume
extracellulaire. Ce déficit est corrigé lorsqu'un traitement à base d'hormone
de croissance est instauré. Une rétention hydrique avec œdème périphérique peut
survenir. Les symptômes sont généralement transitoires, dose-dépendants et
peuvent nécessiter une réduction transitoire de la dose.
Insuffisance
corticosurrénalienne
Une insuffisance corticosurrénalienne a été fréquemment
observée (1,5% dans le GHD pédiatrique et 3 % dans le GHD adulte), voir
rubrique Mises en garde spéciales et précautions d'emploi.
Population pédiatrique
La sécurité de somapacitan a été établie chez les enfants et les adolescents âgés de 3 ans et plus présentant un retard de croissance dû à un GHD. Le profil de sécurité de somapacitan chez les patients présentant un GHD âgés de moins de 3 ans n'a pas été établi.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet: https://signalement.social-sante.gouv.fr.
Envisager une évaluation de l'efficacité et de la sécurité à des intervalles
d'environ 6 à 12 mois.
Pour les enfants, celle-ci peut inclure l'évaluation des paramètres auxologiques, biochimiques (taux d'IGF-1, d'hormones, taux de glucose et de lipides) ainsi que du statut pubertaire. Envisager des évaluations plus fréquentes pendant la puberté.
Pour les adultes, elle peut inclure l'évaluation des paramètres biochimiques (taux d'IGF-1, de glucose et de lipides), de la composition corporelle et de l'indice de masse corporelle.
SURVEILLANCE du traitement :
- Taux de glucose.
- Fonction thyroïdienne.
Effectuer une recherche d'anticorps anti-somapacitan chez les patients ne répondant pas au traitement.
Pour les enfants, celle-ci peut inclure l'évaluation des paramètres auxologiques, biochimiques (taux d'IGF-1, d'hormones, taux de glucose et de lipides) ainsi que du statut pubertaire. Envisager des évaluations plus fréquentes pendant la puberté.
Pour les adultes, elle peut inclure l'évaluation des paramètres biochimiques (taux d'IGF-1, de glucose et de lipides), de la composition corporelle et de l'indice de masse corporelle.
SURVEILLANCE du traitement :
- Taux de glucose.
- Fonction thyroïdienne.
Effectuer une recherche d'anticorps anti-somapacitan chez les patients ne répondant pas au traitement.
CONSULTER le médecin, pharmacien ou infirmier/ère en cas de :
- Maux de tête sévères, problèmes d'acuité visuelle, nausées ou vomissements.
- Douleurs sévères au niveau de l'estomac.
- Douleur persistante à la hanche ou au genou lors de la marche ou une boiterie pendant le traitement.
FEMME en AGE de PROCRÉER : utiliser une méthode de contraception efficace pendant toute la durée du traitement.
SPORTIFS : Substance dopante.
Grossesse
Il n'existe pas de données sur l'utilisation de somapacitan chez la femme enceinte.
Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction, voir rubrique Données de sécurité préclinique.
Sogroya n'est pas recommandé pendant la grossesse et chez les femmes en âge de procréer n'utilisant pas de contraception.
Allaitement
On ne sait pas si somapacitan ou ses métabolites sont excrétés dans le lait maternel.
Les données pharmacologiques/toxicologiques disponibles chez l'animal ont mis en évidence une excrétion de somapacitan dans le lait, voir rubrique Données de sécurité préclinique.
Un risque pour les nouveau-nés ou les nourrissons allaités ne peut être exclu.
Une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre/de s'abstenir de prendre un traitement par Sogroya, en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la femme.
Fertilité
On ne dispose pas d'expérience clinique sur l'utilisation de somapacitan et son effet potentiel sur la fertilité.
Aucun effet indésirable n'a été observé sur la fertilité des mâles et des femelles chez le rat, voir rubrique Données de sécurité préclinique.
Médicaments métabolisés par le cytochrome P450
Les données d'une étude d'interaction réalisée chez des adultes présentant un déficit en hormone de croissance suggèrent que l'administration de l'hormone de croissance peut augmenter la clairance des médicaments connus pour être métabolisés par les isoenzymes du cytochrome P450. La clairance des médicaments métabolisés par le cytochrome P450 (p. ex. les stéroïdes sexuels, les corticostéroïdes, les anticonvulsivants et la ciclosporine) peut en particulier être augmentée, entraînant une diminution des taux plasmatiques de ces médicaments. La conséquence clinique de cet effet n'est pas connue.
Glucocorticoïdes
L'hormone de croissance diminue la conversion de la cortisone en cortisol et peut révéler la présence, jusque-là inconnue, d'un hypoadrénalisme central ou rendre les doses faibles de substitution de glucocorticoïde inefficaces, voir rubrique Mises en garde spéciales et précautions d'emploi.
Œstrogènes par voie orale
Chez les femmes prenant un traitement œstrogénique par voie orale, une dose plus élevée de somapacitan peut être nécessaire pour atteindre l'objectif du traitement, voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi.
Médicaments hypoglycémiants
Un traitement hypoglycémiant, dont l'insuline, peut nécessiter un ajustement de la dose en cas de coadministration avec somapacitan, étant donné que somapacitan peut diminuer la sensibilité à l'insuline, voir rubriques Mises en garde spéciales et précautions d'emploi et Effets indésirables.
Autre
Les effets métaboliques de somapacitan peuvent également être influencés par le traitement concomitant par d'autres hormones, p. ex. la testostérone et les hormones thyroïdiennes, voir rubrique Mises en garde spéciales et précautions d'emploi.
Somapacitan doit être initié et suivi par un médecin spécialisé et expérimenté dans le diagnostic et la prise en charge des patients ayant un déficit en hormone de croissance (p. ex. un endocrinologue).
Posologie
Tableau 1 : Recommandation pour la dose
|
GHD pédiatrique |
Dose initiale recommandée |
|
Patients
pédiatriques naïfs de traitement et patients pédiatriques déjà traités par
d'autres médicaments à base de GH |
0,16 mg/kg/semaine |
|
GHD adulte |
Dose initiale recommandée |
|
Patients naïfs de traitement Adultes (≥ 18 à < 60 ans) Femmes sous traitement
œstrogénique par voie orale (indépendamment de l'âge) Sujets âgés (60 ans et plus)
|
1,5 mg/semaine 2 mg/semaine
1 mg/semaine |
|
Patients déjà traités par des médicaments à base de GH Adultes (≥ 18 à < 60 ans) Femmes sous traitement
œstrogénique par voie orale (indépendamment de l'âge) Sujets âgés (60 ans et plus) |
2 mg/semaine 4 mg/semaine
1,5 mg/semaine
|
GHD pédiatrique
Titration
de la dose
La dose de somapacitan peut être adaptée individuellement
pour chaque patient et ajustée en fonction de la vitesse de croissance, des
effets indésirables, du poids corporel et des concentrations sériques de
facteur de croissance de type insulinique-1 (insulin-like growth factor-1,
IGF-1).
La moyenne des scores de déviation standard (SDS) des taux d'IGF-1 (prélevés 4 jours après l'administration) peut guider la titration de la dose. Les ajustements de dose doivent être considérés pour atteindre une moyenne des scores de déviation standard (SDS) des taux d'IGF-1 dans l'intervalle normal, c'est-à-dire entre -2 et +2 (de préférence proche de 0 SDS).
Si l'IGF-1 (SDS) est > 2, il doit être réévalué après l'administration de la dose suivante de somapacitan. Si la valeur reste > 2, il est recommandé de réduire la dose de 0,04 mg/kg/semaine. Plus d'une réduction de la dose peut être nécessaire chez certains patients.
Chez les patients dont la dose a été réduite mais dont la croissance n'est pas satisfaisante, la dose peut être augmentée progressivement en fonction de la tolérance jusqu'à une dose maximale de 0,16 mg/kg/semaine. Les paliers de dose ne doivent pas dépasser 0,02 mg/kg par semaine.
Évaluation
du traitement
Une évaluation de l'efficacité et de la sécurité doit être
envisagée à intervalles d'environ 6 à 12 mois et peut inclure l'évaluation des
paramètres auxologiques, biochimiques (taux d'IGF-1, d'hormones, taux de
glucose et de lipides) et du statut pubertaire. Des évaluations plus fréquentes
doivent être envisagées pendant la puberté.
Le traitement doit être interrompu chez les patients ayant atteint leur taille définitive ou une taille proche de leur taille définitive, c'est-à-dire avec une vitesse de croissance annualisée < 2 cm/an et un âge osseux > 14 ans chez les filles et > 16 ans chez les garçons, ce qui correspond à la soudure des cartilages de conjugaison épiphysaires, voir rubrique Contre-indications. Une fois les épiphyses soudées, il convient de réévaluer les patients sur le plan clinique afin de déterminer la nécessité ou non d'un traitement par hormone de croissance.
Si le GHD persiste une fois la croissance achevée, le traitement par hormone de croissance devra être maintenu jusqu'au développement somatique complet de l'adulte, incluant la masse corporelle maigre et l'accrétion minérale osseuse (pour des recommandations sur la dose, voir la dose recommandée pour les adultes [tableau 1]).
GHD adulte
Titration
de la dose
La posologie de somapacitan doit être adaptée
individuellement pour chaque patient. Il est recommandé d'augmenter
progressivement la dose à intervalles de 2 à 4 semaines par paliers de 0,5 mg à
1,5 mg en fonction de la réponse clinique des patients et de la survenue
d'effets indésirables jusqu'à une dose de 8 mg de somapacitan par semaine.
Les taux sériques de facteur de croissance de type insulinique-1 (IGF-1) (déterminés entre les jours 3 et 4 après l'administration) peuvent être utilisés comme guide pour la titration de la dose. Le score de déviation standard (SDS) cible d'IGF-1 doit se situer dans l'intervalle normal supérieur sans dépasser 2 SDS. L'intervalle cible des valeurs du SDS IGF-1 est généralement obtenu dans les 8 semaines suivant la titration de la dose. Une titration plus longue de la dose peut être nécessaire chez certains patients adultes présentant un GHD (voir ci-dessous et rubrique Propriétés pharmacodynamiques).
Évaluation
du traitement
En utilisant les valeurs d'IGF-1 (SDS) comme biomarqueur
pour la titration de la dose, l'objectif est d'atteindre des valeurs d'IGF-1
(SDS) comprises dans la partie supérieure de l'intervalle de référence ajusté
en fonction de l'âge (intervalle de référence supérieur IGF-1 (SDS) : 0 à +2)
dans les 12 mois suivant la titration. Si cet intervalle cible ne peut pas être
atteint au cours de cette période ou si le patient n'obtient pas la réponse
clinique souhaitée, d'autres options thérapeutiques doivent être envisagées.
Pendant le traitement d'entretien par somapacitan, une évaluation de l'efficacité et de la sécurité doit être envisagée à intervalles d'environ 6 à 12 mois et peut inclure l'évaluation des paramètres biochimiques (taux d'IGF-1, de glucose et de lipides), de la composition corporelle et de l'indice de masse corporelle.
GHD pédiatrique et adulte
En
remplacement d'autres médicaments à base d'hormone de croissance
Il est recommandé que les patients qui passent d'une
hormone de croissance hebdomadaire au somapacitan poursuivent leur traitement
le même jour que leur jour d'administration hebdomadaire antérieur.
Les patients qui passent d'une hormone de croissance humaine quotidienne au somapacitan une fois par semaine doivent choisir le jour d'administration hebdomadaire qu'ils préfèrent et s'injecter la dernière dose du traitement quotidien le jour précédant (ou au moins 8 heures avant) l'injection de la première dose de somapacitan une fois par semaine. Les patients doivent suivre les instructions relatives à la dose présentées dans le tableau 1.
Traitement
œstrogénique par voie orale
Les femmes prenant un traitement œstrogénique par voie
orale peuvent présenter des taux d'IGF-1 réduits et nécessiter un ajustement de
la dose en hormone de croissance pour atteindre l'objectif du traitement (voir
rubrique Mises en garde spéciales et précautions d'emploi).
Dans le cadre du GHD pédiatrique, des doses supérieures à 0,16 mg/kg/semaine n'ont pas été étudiées et ne sont pas recommandées.
Oubli
de dose
Il est conseillé aux patients qui oublient une dose de
prendre somapacitan en injection hebdomadaire dès qu'ils s'en rendent compte,
dans les 3 jours après la dose oubliée, puis de reprendre leur schéma
posologique habituel d'une fois par semaine. Si plus de 3 jours se sont
écoulés, la dose ne doit pas être administrée, et la dose suivante doit être
administrée le jour normalement prévu. Si deux ou plusieurs doses ont été
oubliées, la dose doit être reprise le jour normalement prévu.
Modification
du jour d'administration
Le jour de l'injection hebdomadaire peut être changé, à
condition que le délai entre deux doses soit d'au moins 4 jours. Après avoir
choisi un nouveau jour d'administration, il faut continuer d'administrer la
dose une fois par semaine.
Flexibilité
du jour d'administration
Dans les cas où l'injection de la dose n'est pas possible
le jour d'administration prévu, somapacitan une fois par semaine peut être
administré au maximum 2 jours avant ou 3 jours après le jour d'administration hebdomadaire prévu, à condition que le
délai entre deux doses soit d'au moins 4 jours (96 heures). L'administration
une fois par semaine pour la dose suivante peut être reprise le jour
d'administration normalement prévu.
Populations particulières
Sujets
âgés (60 ans et plus)
Généralement, des doses plus faibles de somapacitan peuvent
être nécessaires chez les patients plus âgés. Pour des informations
supplémentaires, voir rubrique Propriétés pharmacocinétiques.
Population
pédiatrique
Il existe des données limitées sur les effets cliniques de
somapacitan chez les patients pédiatriques présentant un GHD âgés de moins de 3
ans. Les données actuellement disponibles sont décrites en rubrique Propriétés pharmacodynamiques et Propriétés pharmacocinétiques,
mais aucune recommandation sur une posologie ne peut être émise.
Sexe
Les hommes présentent une augmentation de la sensibilité à
l'IGF-1 au cours du temps. Cela signifie qu'il existe un risque que les hommes
soient surdosés. Les femmes, en particulier celles prenant des œstrogènes par
voie orale, peuvent nécessiter des doses plus élevées et une période de
titration plus longue que les hommes, voir rubriques Propriétés pharmacodynamiques et Propriétés pharmacocinétiques. Chez les
femmes sous œstrogène par voie orale, il faut envisager de changer la voie
d'administration de l'œstrogène (p. ex. voie transdermique, vaginal), voir rubrique
Mises en garde spéciales et précautions d'emploi.
Insuffisance
rénale
Aucun ajustement de la dose initiale n'est nécessaire chez
les patients présentant une insuffisance rénale. Les patients présentant une
insuffisance rénale peuvent nécessiter des doses plus faibles de somapacitan,
mais comme la dose de somapacitan est ajustée individuellement en fonction des
besoins de chaque patient, aucun ajustement supplémentaire de la dose n'est
nécessaire (voir rubrique Propriétés pharmacocinétiques).
Insuffisance
hépatique
Aucun ajustement de la dose initiale n'est nécessaire chez
les patients présentant une insuffisance hépatique. Les patients présentant une
insuffisance hépatique modérée peuvent nécessiter des doses plus élevées de
somapacitan, mais comme la dose de somapacitan est ajustée individuellement en
fonction des besoins de chaque patient, aucun ajustement supplémentaire de la
dose n'est nécessaire. On ne dispose d'aucune information concernant
l'utilisation de somapacitan chez les patients présentant une insuffisance
hépatique sévère. Il convient d'être prudent lors du traitement de ces patients
avec somapacitan, voir rubrique Propriétés pharmacocinétiques.
Mode d'administration
Somapacitan doit être administré une fois par semaine, quel que soit le moment de la journée.
Somapacitan doit être injecté par voie sous-cutanée dans l'abdomen, les cuisses, les fesses ou les hauts du bras, sans ajustement de la dose.
Le site d'injection doit être changé chaque semaine afin d'éviter une lipoatrophie locale.
Sogroya 10 mg/1,5 ml, solution injectable en stylo prérempli
Le stylo Sogroya 10 mg/1,5 ml (6,7 mg/ml) délivre des doses de 0,05 mg (0,0075 ml) à 4 mg (0,6 ml) par paliers de 0,05 mg.
Pour les précautions à prendre avant l'administration du médicament, voir la rubrique Précautions particulières d'élimination et de manipulation.
Durée de conservation :
2 ans.
Après la première
ouverture
6 semaines au réfrigérateur (entre 2 °C et 8 °C).
Ne pas congeler. Maintenir à distance de l'élément de refroidissement.
Conservez Sogroya dans l'emballage extérieur avec le capuchon du stylo pour le protéger de la lumière.
Avant et après la
première ouverture
Si une conservation au réfrigérateur n'est pas possible
(par exemple en cas de déplacement), Sogroya peut être conservé temporairement
à une température ne dépassant pas 30 °C pendant une durée totale de 72 heures
(3 jours). Remettre Sogroya au réfrigérateur après conservation à cette
température. En cas de conservation en dehors du réfrigérateur puis retour de
nouveau au réfrigérateur, la durée totale de conservation en dehors du
réfrigérateur ne doit pas dépasser 3 jours, surveiller ceci attentivement. Le
stylo Sogroya doit être jeté s'il a été conservé à une température ne dépassant
pas 30 °C pendant plus de 72 heures (3 jours) ou pour toute période de temps,
conservé à une température supérieure à 30 °C.
Précautions particulières de conservation :
À conserver au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler. Maintenir à distance de l'élément de refroidissement.
Conserver Sogroya dans l'emballage extérieur avec le capuchon du stylo pour le protéger de la lumière.
Pour les conditions de conservation du médicament après première ouverture, voir la rubrique Durée de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Il existe une expérience clinique limitée du surdosage de somapacitan.
D'après l'expérience acquise avec le traitement quotidien à base d'hormone de croissance, un surdosage à court terme peut entraîner initialement une diminution de la glycémie, suivie d'une augmentation de la glycémie. Cette diminution de la glycémie a été détectée à l'analyse biochimique mais sans signes cliniques d'hypoglycémie.
Un surdosage à long terme pourrait entraîner des signes et des symptômes correspondant aux effets connus d'un excès en hormone de croissance humaine.
Classe pharmacothérapeutique : Hormones hypophysaires, hypothalamiques et analogues, somatropine et agonistes de la somatropine, Code ATC : H01AC07.
Mécanisme d'action
Somapacitan est un dérivé recombinant de l'hormone de croissance humaine à longue durée d'action. Il est constitué de 191 acides aminés similaires à l'hormone de croissance humaine endogène, avec une substitution unique dans le squelette d'acides aminés (L101C) auquel une fraction de liaison à l'albumine a été attachée. La fraction de liaison à l'albumine (chaîne latérale) consiste en une fraction d'acide gras et un espaceur hydrophile attachés en position 101 de la protéine.
Somapacitan agit soit directement via le récepteur GH et/ou indirectement via l'IGF-1 produit dans les tissus de l'organisme, mais essentiellement dans le foie.
Lorsque le déficit en hormone de croissance est traité par somapacitan, une normalisation de la composition corporelle (c.-à-d. une réduction de la masse grasse corporelle, une augmentation de la masse maigre) et de l'action métabolique est obtenue.
Somapacitan stimule la croissance osseuse chez les patients pédiatriques présentant un GHD en raison de ses effets sur les cartilages de conjugaison (épiphyses) des os, voir rubrique Données de sécurité préclinique.
Effets pharmacodynamiques
IGF-1
L'IGF-1 est un biomarqueur généralement reconnu de
l'efficacité dans le GHD.
Une réponse d'IGF-1 dose-dépendante est induite après
l'administration de somapacitan.
L'état d'équilibre des réponses d'IGF-1 est atteint après 1
à 2 doses hebdomadaires.
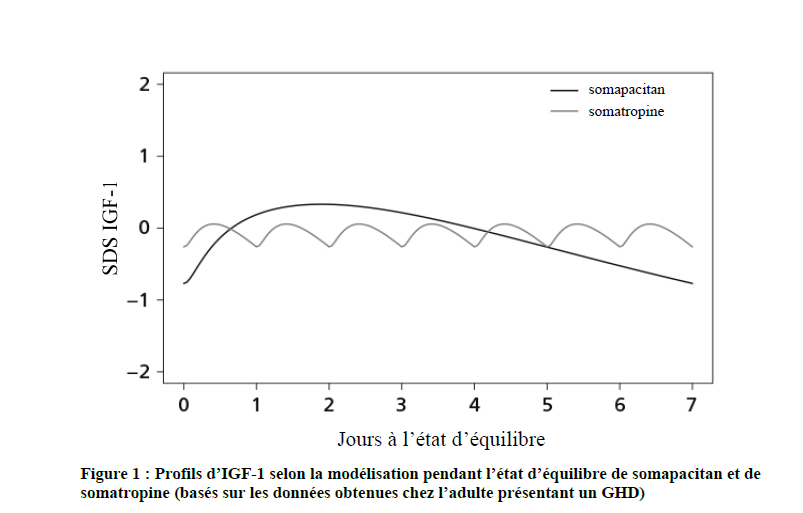
Les taux d'IGF-1 fluctuent au cours de la semaine. La réponse d'IGF-1 est maximale au bout de 2 à 4 jours. Comparativement au traitement quotidien par la GH, le profil d'IGF-1 de somapacitan est différent, voir Figure 1.
Chez les patients présentant un GHD pédiatrique, somapacitan produit une réponse de l'IGF-1 dose- linéaire, un changement de dose de 0,02 mg/kg en moyenne entraînant un changement du score de déviation standard (SDS) de l'IGF-1 de 0,32.
Efficacité et sécurité cliniques
GHD pédiatrique
REAL
4 (phase 3)
L'efficacité et la sécurité de somapacitan une fois par semaine ont été évaluées dans un essai de phase 3 multicentrique, randomisé, en ouvert, contrôlé versus un médicament actif, en groupes parallèles, d'une durée de 52 semaines (REAL 4) chez 200 patients pédiatriques naïfs de traitement présentant un GHD. Les patients ont été randomisés pour recevoir 0,16 mg/kg/semaine de somapacitan une fois par semaine (N = 132) ou 0,034 mg/kg/jour de somatropine une fois par jour (N = 68).
À l'inclusion, les 200 patients avaient un âge moyen de 6,4 ans (intervalle de 2,5 à 11 ans). 74,5 % des patients étaient de sexe masculin.
La vitesse de croissance annualisée à la semaine 52 était similaire pour somapacitan et somatropine (tableau 4).
Tableau 4 :
Résultats de croissance à la semaine 52 chez les patients pédiatriques
présentant un GHD
|
|
Somapacitan une fois par semaine (N = 132) |
Somatropine
une fois par jour (N = 68) |
Estimation
de la différence entre les traitements (IC à 95 %) (somapacitan moins somatropine) |
|
Vitesse de croissance annualisée (cm/an) |
11,2 |
11,7 |
-0,5 [-1,1 ; 0,2] |
En accord avec cela, les variations du SDS de la taille et du SDS de l'IGF-1 à la semaine 52 par rapport à l'inclusion étaient également similaires entre somapacitan et somatropine (tableau 5).
Tableau 5 :
Déviations standard de la taille et de l'IGF-1 chez les patients pédiatriques
présentant un GHD — traitement de 52 semaines
|
|
Somapacitan une fois par semaine (N = 132) |
Somatropine une fois par jour (N = 68) |
Estimation
de la différence entre les traitements (IC à 95 %) (somapacitan moins somatropine) |
|
Déviation standard de la Taille
(SDS) à l'inclusiona |
-2,99 |
-3,47 |
|
|
Déviation
standard de la taille (SDS) : variation par rapport à l'inclusion |
1,25 |
1,30 |
-0,05 [-0,18 ; 0,08] |
|
Déviation
standard du taux d'IGF-1 (SDS), à l'inclusiona |
-2,03 |
-2,33 |
|
|
Déviation standard des taux d'IGF-1 (SDS), à la Semaine 52a |
0,28 |
0,10 |
|
|
Déviation
standard des taux d'IGF-1 (SDS) variation par rapport à l'inclusion |
2,36 |
2,33 |
0,03 [-0,30 ; 0,36] |
a Moyenne observée
La grande majorité des patients pédiatriques (96,9 %) de l'essai ont obtenu une moyenne des scores de déviation standard (SDS) des taux d'IGF-1 dans l'intervalle normal (-2 à +2) après 52 semaines de traitement par somapacitan une fois par semaine (tableau 6). Un faible nombre de patients avait un SDS de l'IGF-1 moyen supérieur à +2 (2,3 %) et aucun patient n'avait une moyenne des scores de déviation standard (SDS) des taux d'IGF-1 supérieure à +3.
Tableau 6 :
Moyenne des valeurs des scores de déviations standard (SDS) des taux d'IGF-1 après 52
semaines de traitement de patients pédiatriques présentant un GHD par
somapacitan une fois par semaine
|
Catégorie du SDS de l'IGF-1 |
Moyenne à la semaine 52
(N = 132) |
|
< -2 |
0,8 % |
|
-2 à 0 |
21,2 % |
|
0 à +2 |
75,8 % |
|
+2 à +3 |
2,3 % |
|
> +3 |
0 |
REAL
3 (phase 2)
Au total, 59 patients pédiatriques naïfs de
traitement avec un GHD ont terminé une période principale de 26 semaines et une
phase d'extension de 26 semaines dans un essai en groupes parallèles à 4 bras
avec le somapacitan administré une fois par semaine à des doses de 0,04, 0,08
et 0,16 mg/kg/semaine et la somatropine administrée quotidiennement à la dose
de 0,034 mg/kg/jour dans le cadre d'un bras témoin actif. Les patients ont
continué dans une phase d'extension de sécurité en ouvert de 104 semaines, en groupes
parallèles, avec somapacitan0,16 mg/kg/semaine et somatropine
quotidienne 0,034 mg/kg/jour. Tous les patients ont ensuite été transférés au
somapacitan une fois par semaine 0,16 mg/kg/semaine dans le cadre d'une phase
d'extension de sécurité à long terme de 208 semaines.
Le traitement par somapacitan administré une fois par semaine a permis d'obtenir des bénéfices thérapeutiques continus au moins jusqu'à la semaine 208. Le score de déviation standard (SDS) de la taille était de -1,06 (variation par rapport à l'inclusion : 2,85) chez 38 patients.
Les valeurs de la taille obtenues à la semaine 208 chez les patients passés de 0,034 mg/kg/jour de la somatropine administrée quotidiennement à 0,16 mg/kg/semaine de somapacitan administré une fois par semaine à la semaine 156, indiquent que les bénéfices du traitement quotidien par GH sont maintenus après passage au somapacitan administré une fois par semaine.
Les valeurs moyennes des scores de déviations standard (SDS) des taux d'IGF-1 sont restées dans l'intervalle normal pour tous les groupes.
GHD adulte
Dans un essai de 34 semaines contrôlé versus placebo
(en double aveugle) et contrôlé versus
un médicament actif (en ouvert),
301 patients adultes naïfs de traitement avec un GHD ont été randomisés
(2:1:2) dont 300 ont été exposés au somapacitan ou à un placebo une
fois par semaine ou à
somatropine administrée de façon quotidienne pendant une période de
traitement
de 34 semaines (phase principale de l'étude). La population de patients
avait
un âge moyen de 45,1 ans (intervalle de 23 à 77 ans ; 41 patients
étaient âgés
de 65 ans ou plus), 51,7 % étaient des femmes et 69,7 % avaient un GHD
apparu à
l'âge adulte.
Un total de 272 patients adultes présentant un GHD ayant
terminé la phase principale de 34 semaines ont continué dans une phase
d'extension en ouvert de 53 semaines. Les sujets sous placebo sont passés au
somapacitan et les patients sous somatropine ont été à nouveau randomisés (1:1)
pour recevoir somapacitan ou somatropine.
Les effets cliniques observés pour les critères principaux d'évaluation dans la phase de traitement principale (tableau 7) et la phase d'extension du traitement (tableau 8) sont présentés ci-dessous.
Tableau 7 : Résultats à 34 semaines|
Variation entre l'inclusion et la semaine 34a |
somapacitan |
somatropine |
placebo |
Différence somapacitan- placebo [IC 95 %] valeur de p |
Différence somapacitan- somatropine [IC 95 %] |
|
Nombre de sujets (N) |
120 |
119 |
61 |
|
|
|
% graisse tronculaire (Critère principal)
|
-1,06 |
-2,23 |
0,47 |
-1,53 [-2,68 ; -0,38] 0,0090b |
1,17 [0,23 ; 2,11] |
|
Tissu adipeux viscéral (cm2) |
-10 |
-9 |
3 |
-14 [-21 ; -7]
|
-1 [-7 ; 4]
|
|
Masse musculaire appendiculaire (g) |
558 | 462 | -121 |
679
[340 ; 1 019]
|
96 [-182 ; 374]
|
|
Masse corporelle |
1 394 | 1 345 | 250 |
1 144
[459 ; 1 829]
|
49 [-513 ; 610]
|
|
SDS du taux d'IGF-1 |
2,40 |
2,37 |
-0,01 |
2,40 [2,09 ; 2,72]
|
0,02 [-0,23 ; 0,28]
|
Abréviations : N = nombre de sujets dans la
population totale d'analyse, IC = intervalle de confiance. SDS IGF-1 : score de déviation standard du
facteur de croissance de type insulinique-1
a Les paramètres de composition corporelle sont basés sur l'absorptiométrie biphotonique à rayons X (DXA).
b L'analyse principale était une comparaison par rapport aux valeurs basales des variations en % de la graisse tronculaire avec somapacitan et avec le placebo. Les variations en % de la graisse tronculaire entre les valeurs basales et celles de la semaine 34 ont été analysées en utilisant un modèle d'analyse de covariance avec le traitement, le type de survenue du GHD, le sexe, la région, la présence d'un diabète sucré et l'interaction sexe par région par présence d'un diabète sucré comme facteurs et la valeur basale comme covariable en intégrant une technique d'imputation multiple où les valeurs manquantes de la semaine 34 ont été imputées sur la base des données du groupe placebo.
a Les paramètres de composition corporelle sont basés sur l'absorptiométrie biphotonique à rayons X (DXA).
b L'analyse principale était une comparaison par rapport aux valeurs basales des variations en % de la graisse tronculaire avec somapacitan et avec le placebo. Les variations en % de la graisse tronculaire entre les valeurs basales et celles de la semaine 34 ont été analysées en utilisant un modèle d'analyse de covariance avec le traitement, le type de survenue du GHD, le sexe, la région, la présence d'un diabète sucré et l'interaction sexe par région par présence d'un diabète sucré comme facteurs et la valeur basale comme covariable en intégrant une technique d'imputation multiple où les valeurs manquantes de la semaine 34 ont été imputées sur la base des données du groupe placebo.
L'analyse post hoc en sous-groupes des variations en pourcentage (%) de la graisse tronculaire versus placebo entre les valeurs basales et celles de la semaine 34 a montré une différence entre traitements (somapacitan-placebo) estimée de -2,49 % [-4,19 ; -0,79] chez les hommes, -0,80 % [-2,99 ; 1,39] chez les femmes ne recevant pas d'œstrogène par voie orale, -1,44 % [-3,97 ; 1,09] chez les femmes prenant un œstrogène par voie orale.
Tableau 8 : Résultats à 87 semaines|
Variation entre l'inclusion et la semaine 87a |
somapacitan/ somapacitan |
somatropine/ somatropine
| placebo/
somapacitan |
somatropine/ somapacitan |
Différence somapacitan/
somapacitan vs somatropine/somatropine [IC%] |
|
Nombre de sujets (N) |
114 |
52 |
54 |
51 |
|
|
% graisse tronculaire |
-1,52 |
-2,67 |
-2,28 |
-1,35 |
1,15 [-0,10 ; 2,40]
|
|
Tissu adipeux viscéral (cm2) |
-6,64 |
-6,85 |
-10,21 |
-8,77 |
0,22 [-10 ; 10]
|
|
Masse musculaire squelettique appendiculaire (g) |
546,11 |
449,09 |
411,05 |
575,80 |
97,02 [-362 ; 556]
|
|
Masse corporelle maigre (g) |
1 739,05 | 1 305,73 | 1 660,56 | 1 707,82 |
433,32
[-404 ; 1 271]
|
a Les paramètres de composition corporelle sont basés
sur l'imagerie DXA.
Valeurs SDS IGF-1 observées et simulées dans l'étude clinique
Dans la phase principale de l'étude clinique, des valeurs SDS IGF-1 de 0 et plus ont globalement été obtenues chez 53 % des patients adultes de l'étude présentant un GHD traités par somapacitan après une période de titration de la dose de 8 semaines. Cette proportion était toutefois plus faible dans des sous-groupes particuliers tels que les femmes sous œstrogène par voie orale (32 %) et les patients avec un déficit acquis dans l'enfance (39 %) (Tableau 9). Les analyses de simulation post hoc ont indiqué que les proportions de patients adultes présentant un GHD obtenant des valeurs SDS IGF-1 supérieures à 0 devraient être plus élevées si la titration de la dose de somapacitan au-delà de 8 semaines était autorisée. Dans cette analyse de simulation, il a été supposé que la titration de la dose de somapacitan ayant bien tolérée chez tous les patients qui ont atteint des valeurs SDS IGF-1 situées dans l'intervalle cible ou avec une dose de somapacitan de 8 mg par semaine.
Tableau 9 Proportions de patients adultes présentant un GHD traités par somapacitan avec des valeurs SDS IGF-1 supérieures à 0|
Sous-groupes |
Hommes |
Femmes ne prenant pas d'œstrogène oral |
Femmes prenant un œstrogène oral |
GHD adulte acquis dans l'enfance
|
GHD adulte apparu à l'âge adulte |
Total |
|
Observéesa |
71 % |
46 % |
32 % |
39 % |
60 % |
53 % |
|
Simulations post hoc |
100 % |
96 % |
70 % |
84 % |
92 % |
90 % |
Dose d'entretien
La dose d'entretien varie d'une personne à l'autre et
entre les hommes et les femmes. La dose d'entretien moyenne de somapacitan
observée dans les essais cliniques de phase 3 était de 2,4 mg/semaine.
GHD pédiatrique
et adulte
Sécurité clinique
Le profil de sécurité de somapacitan a été similaire au
profil de sécurité bien connu de somatropine. Aucun nouveau problème de
sécurité n'a été identifié, voir rubrique Effets indésirables.
Immunogénicité
Les anticorps anti-médicament (Anti-drug antibodies, ADA)
ont été rarement détectés chez les patients pédiatriques (16/132). Aucun de ces
anticorps n'était neutralisant. Aucune preuve de l'impact des ADA sur la
pharmacocinétique, l'efficacité ou la sécurité n'a été observée. Aucun
anticorps antimédicament n'a été détecté chez les patients adultes.
Population
pédiatrique
L'Agence Européenne des Médicaments a renoncé à
l'obligation de soumettre les résultats d'études réalisées avec Sogroya dans
tous les sous-groupes de la population pédiatrique dans le déficit en hormone
de croissance (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage
pédiatrique).
Somapacitan a des propriétés pharmacocinétiques compatibles
avec l'administration une fois par semaine. La liaison réversible à l'albumine
endogène retarde l'élimination de somapacitan et prolonge ainsi la demi-vie in
vivo et la durée d'action.
La pharmacocinétique de somapacitan après l'administration
sous-cutanée a été évaluée à des doses de 0,02 à 0,16 mg/kg/semaine dans la population
pédiatrique (2,5 à 14 ans), à des doses de 0,01 à 0,32 mg/kg chez des adultes
sains et à des doses allant jusqu'à 0,12 mg/kg chez des patients adultes
présentant un GHD.
Globalement, somapacitan présente une pharmacocinétique non
linéaire dans l'intervalle de doses étudiées. En revanche, dans l'intervalle de
doses cliniquement pertinentes de somapacitan chez les adultes présentant un
GHD, la pharmacocinétique de somapacitan est approximativement linéaire.
Dans le GHD pédiatrique, une dose de somapacitan de 0,16 mg/kg/semaine correspond à une concentration moyenne de 80,2 ng/ml et, dans le GHD adulte, les doses de somapacitan dans l'intervalle de doses cliniquement pertinentes correspondent à des concentrations moyennes de 0,1 ng/ml à 36,2 ng/ml.
Absorption
Chez les patients adultes et pédiatriques présentant un
GHD, le tmax médian allait de 4 à 25,5 heures à des doses allant de
0,02 mg/kg/semaine à 0,16 mg/kg/semaine.
L'exposition à l'état d'équilibre a été atteinte
après une administration 1-2 fois par semaine. La biodisponibilité absolue de
somapacitan chez les humains n'a pas été étudiée.
Distribution
Somapacitan est fortement lié (> 99 %) aux protéines plasmatiques et devrait être distribué de manière identique à l'albumine. D'après les analyses de PK de population, le volume de distribution estimé (V/F) était de 1,7 l chez les patients pédiatriques présentant un GHD et de 14,6 l chez les patients adultes présentant un GHD.
Élimination
Après l'administration d'une dose unique et de doses répétées de 0,16 mg/kg/semaine, la demi-vie terminale était d'environ 34 heures chez les patients pédiatriques présentant un GHD.
La demi-vie terminale a été estimée avec des moyennes géométriques d'environ 2 à 3 jours à l'état d'équilibre chez les patients pédiatriques et adultes présentant un GHD (doses : 0,02 à 0,12 mg/kg). Somapacitan sera présent dans la circulation pendant environ 2 semaines après la dernière dose. Une accumulation faible à nulle (rapport moyen d'accumulation : 1-2) de somapacitan après des administrations répétées a été observée.
Biotransformation
Somapacitan est fortement métabolisé par dégradation protéolytique et clivage de la séquence de liaison entre le peptide et le motif liant l'albumine.
Somapacitan était fortement métabolisé avant l'excrétion et il n'a pas été retrouvé de somapacitan intact ni dans l'urine, qui était la principale voie d'excrétion (81 %), ni dans les fèces où 13 % des produits liés au somapacitan étaient retrouvés, indiquant une complète biotransformation avant l'excrétion.
Populations particulières
Patients pédiatriques
présentant un GHD
D'après une analyse pharmacocinétique de population, le
sexe, l'origine ethnique et le poids corporel n'ont pas d'effet cliniquement
significatif sur la pharmacocinétique après une administration en fonction du
poids.
Patients adultes
présentant un GHD
Âge
Les sujets de plus de 60 ans ont une exposition supérieure
(29 %) par rapport aux sujets plus jeunes pour une même dose de somapacitan.
Une dose initiale plus faible pour les sujets de plus de 60 ans est décrite à
la rubrique Posologie et mode d'administration.
Sexe
Les femmes, et en particulier les femmes prenant un
œstrogène oral, ont une exposition plus faible (53 % pour les femmes sous
œstrogène oral et 30 % pour les femmes sans œstrogène oral) que les hommes pour
la même dose de somapacitan. Une dose initiale plus élevée pour les femmes
prenant un œstrogène oral est décrite dans la rubrique Posologie et mode d'administration.
Origine
ethnique
Il n'y avait pas de différence d'exposition au somapacitan
et de réponse d'IGF-1 entre les sujets japonais et caucasiens. Malgré une
exposition plus élevée chez les Asiatiques non japonais par rapport aux
caucasiens à la même dose de somapacitan, les caucasiens, les japonais et les
Asiatiques non japonais avaient besoin des mêmes doses pour atteindre des taux
similaires d'IGF-1. Par conséquent, il n'y a pas de recommandation d'ajustement
de la dose en fonction de l'origine ethnique.
Ethnicité
L'ethnicité (hispanique ou latino 4,5 % (15 sujets ont reçu
somapacitan)) n'a pas été évaluée en raison de la taille réduite de
l'échantillon dans le programme de développement.
Poids
corporel
Malgré une exposition plus élevée chez les sujets à faible poids
corporel par rapport aux sujets à poids corporel élevé pour la même dose de
somapacitan, les sujets avaient besoin des mêmes doses pour atteindre des taux
d'IGF-1 similaires dans l'intervalle de poids corporels allant de 35 kg à 150
kg. Par conséquent, il n'y a pas de recommandation d'ajustement de la dose en
fonction du poids corporel.
Insuffisance
rénale
Une dose de somapacitan de 0,08 mg/kg à l'état d'équilibre
s'est traduite par des expositions plus élevées chez les sujets insuffisants
rénaux, plus prononcées chez les sujets ayant une insuffisance rénale sévère et
chez les sujets nécessitant une hémodialyse, pour lesquels les rapports des ASC
0-168h de la fonction rénale normale étaient respectivement de 1,75
et 1,63. En général, l'exposition au somapacitan avait tendance à augmenter
avec la diminution du DFG.
Des taux d'IGF-I ASC0-168h plus élevés ont été
observés chez les sujets présentant une insuffisance rénale modérée et sévère
et chez les sujets nécessitant une hémodialyse, avec des ratios relatifs à la
fonction rénale normale de respectivement 1,35, 1,40 et 1,24.
En raison de la modeste augmentation observée en IGF-1
combinée aux faibles doses initiales recommandées et la titration individuelle
de la dose de somapacitan, il n'y a pas de recommandation d'ajustement de la
dose chez les patients insuffisants rénaux.
Insuffisance
hépatique
Une dose de somapacitan de 0,08 mg/kg à l'état d'équilibre
s'est traduite par une exposition plus élevée chez les sujets atteints
d'insuffisance hépatique modérée avec des rapports relatifs à la fonction
hépatique normale de 4,69 pour l'ASC0-168h et de 3,52 pour la Cmax.
Des taux plus faibles d'IGF-1 stimulé par somapacitan ont
été observés chez les sujets atteints d'insuffisance hépatique légère et modérée
par rapport aux sujets ayant une fonction hépatique normale (les ratios
relatifs à la fonction hépatique normale étaient de 0,85 pour l'insuffisance
hépatique légère et de 0,75 pour l'insuffisance hépatique modérée).
En raison de la diminution modeste observée en IGF-1
combinée à l'ajustement individuel de la dose de somapacitan, il n'y a pas de
recommandation d'ajustement de la dose chez les patients insuffisants
hépatiques.
Sogroya n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Les données précliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration répétée, génotoxicité ou de développement pré- et postnatal ne révèlent pas de risque particulier pour l'homme.
Aucune étude de carcinogénicité n'a été réalisée avec somapacitan.
Aucun effet indésirable n'a été observé sur la fertilité des mâles et des femelles chez le rat à des doses se traduisant par une exposition au moins 13 et 15 fois plus élevée que l'exposition clinique maximale attendue à 8 mg/semaine pour les mâles et les femelles, respectivement. Toutefois, un cycle œstral irrégulier chez la femelle a été observé à toutes les doses étudiées.
Aucune preuve d'atteinte fœtale n'a été
identifiée lorsque somapacitan a été administré à des rates et des
lapines gestantes pendant
l'organogenèse à des doses se traduisant par des expositions nettement
supérieures à l'exposition attendue pour la dose clinique maximale de 8
mg/semaine (au moins 18 fois). Aux doses élevées entraînant une
exposition au
moins 130 fois supérieure à l'exposition clinique maximale attendue à 8
mg/semaine, un raccourcissement/une courbure/un épaississement des os
longs ont
été observés chez les petits des rates recevant somapacitan. De tels
résultats
chez le rat sont connus pour disparaître après la naissance et doivent
être
considérés comme des malformations mineures, non comme des anomalies
permanentes.
La croissance fœtale était ralentie lorsque somapacitan
était administré par voie sous-cutanée à des lapines gestantes à des
expositions au moins 9 fois supérieures à l'exposition attendue pour la dose
clinique maximale de 8 mg/semaine.
Chez des rates allaitantes, des produits liés au somapacitan étaient sécrétés dans le lait mais à un niveau inférieur à celui observé dans le plasma (jusqu'à 50 % du taux plasmatique).
Le stylo est réservé à l'utilisation par un seul patient.
Sogroya ne doit pas être utilisé si la solution n'est pas limpide à légèrement opalescente, incolore à légèrement jaune et exempte de particules visibles.
Sogroya ne doit pas être utilisé s'il a été congelé.
La cartouche ne doit pas être retirée du stylo prérempli et remplie à nouveau.
Une aiguille doit toujours être fixée avant l'utilisation. Les aiguilles ne doivent pas être réutilisées. L'aiguille d'injection doit être retirée après chaque injection et le stylo doit être conservé sans aiguille fixée. Cela pourra prévenir le risque d'obstruction des aiguilles, de contamination, d'infection, de fuite de la solution et de dose incorrecte.
En cas d'aiguilles obstruées, les patients doivent suivre les instructions décrites dans les instructions d'utilisation accompagnant la notice.
Les aiguilles ne sont pas incluses. Le stylo prérempli Sogroya est conçu pour être utilisé avec des aiguilles à usage unique d'une longueur comprise entre 4 mm et 8 mm et d'un diamètre compris entre 30 G et 32 G.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
liste I
Prescription initiale hospitalière annuelle.
Prescription initiale hospitalière annuelle.
Prescription initiale et renouvellement réservés aux spécialistes en pédiatrie ou en endocrinologie-diabétologie-nutrition.
Médicament d'exception : prescription en conformité avec la fiche d'information thérapeutique.
Médicament d'exception : prescription en conformité avec la fiche d'information thérapeutique.
Solution injectable (injection).
Liquide limpide à légèrement opalescent, incolore à
légèrement jaune et exempt de particules visibles.
Le stylo prérempli contient 1,5 ml de solution dans une cartouche en verre (verre incolore de Type I) avec un piston en caoutchouc chlorobutyle et un bouchon en caoutchouc bromobutyle/isoprène scellé avec une capsule en aluminium. La cartouche est contenue dans un stylo multidoses jetable en polypropylène, polyacétal, polycarbonate et acrylonitrile butadiène styrène, avec en plus deux ressorts métalliques. La cartouche est scellée de manière permanente dans un stylo prérempli.
Sogroya 10 mg/1,5 ml, solution injectable en stylo prérempli
Stylo prérempli à code couleur dont le bouton de dose du stylo est de couleur jaune.
Boîte de 1 stylo prérempli.
Sogroya 10 mg/1,5 ml, solution injectable en stylo prérempli
Un ml de solution contient 6,7 mg de somapacitan*
Chaque stylo prérempli contient 10 mg de somapacitan dans 1,5 ml de solution
*Produit par la technologie de l'ADN recombinant dans Escherichia coli suivie par l'attachement d'une fraction de liaison à l'albumine.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Histidine
Mannitol
Poloxamère 188
Phénol
Eau pour préparations injectables
Acide chlorhydrique (pour l'ajustement du pH)
Hydroxyde de sodium (pour l'ajustement du pH)
Commercialisation de SOGROYA 10 mg/1,5 ml, solution injectable en stylo prérempli, boîte de 1 stylo prérempli multidose de 1,50 ml (34009 3024729 7) le 25/08/2025