
BUGVI 5 mg-mL, poudre pour dispersion pour perfusion, boîte de 1 flacon de 100 mg
Dernière révision : 03/12/2024
Taux de TVA : 2.1%
Laboratoire exploitant : EG LABO
Source :

BUGVI est indiqué en monothérapie dans le traitement du cancer du sein métastatique chez les patients adultes en échec du traitement de première ligne du cancer métastatique et pour qui le traitement conventionnel incluant une anthracycline n'est pas indiqué (voir rubrique Mises en garde spéciales et précautions d'emploi).
BUGVI en association avec la gemcitabine est indiqué dans le traitement de première ligne de l'adénocarcinome du pancréas métastatique chez les patients adultes.
BUGVI en association avec le carboplatine est indiqué dans le traitement de première ligne du cancer bronchique non à petites cellules chez les patients adultes qui ne sont pas candidats à une chirurgie potentiellement curative et/ou une radiothérapie.
- Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
- Allaitement (voir rubrique Fertilité, grossesse et allaitement).
- Patients dont le taux de neutrophiles à l'inclusion est < 1 500/mm3.
BUGVI est une formulation de nanoparticules de paclitaxel-albumine pouvant avoir des propriétés pharmacologiques très différentes des autres formulations de paclitaxel (voir rubriques Propriétés pharmacodynamiques et Propriétés pharmacocinétiques). Elle ne doit pas être remplacée par d'autres formulations de paclitaxel et elle ne doit pas être utilisée à leur place.
Hypersensibilité
De rares cas de réaction d'hypersensibilité sévères, dont de très rares cas de réactions anaphylactiques d'issue fatale, ont été rapportés. En cas de réaction d'hypersensibilité, le médicament doit être immédiatement arrêté, un traitement symptomatique doit être mis en place et le patient ne doit pas être à nouveau exposé au paclitaxel.
Hématologie
Une aplasie médullaire (principalement une neutropénie) survient fréquemment avec le paclitaxel sous forme de nanoparticules liées à l'albumine. La neutropénie est dose-dépendante et constitue une toxicité dose-limitante. Une surveillance fréquente de la numération sanguine doit être effectuée tout au long du traitement par BUGVI. Il convient de reprendre le traitement par BUGVI uniquement si le taux de neutrophiles redevient > 1 500/mm3 et le taux de plaquettes > 100 000/mm3 (voir rubrique Posologie et mode d'administration).
Neuropathie
Les neuropathies sensitives sont fréquentes avec le paclitaxel sous forme de nanoparticules liées à l'albumine. Toutefois, des symptômes sévères se développent moins fréquemment. La survenue d'une neuropathie sensitive de grade 1 ou 2 ne nécessite généralement pas de réduction de dose. En cas d'apparition d'une neuropathie sensitive de grade 3 pendant le traitement par BUGVI en monothérapie, le traitement doit être suspendu jusqu'à la régression de la neuropathie à un grade 1 ou 2, et il est alors recommandé de réduire la dose pour toutes les cures suivantes de BUGVI (voir rubrique Posologie et mode d'administration). En cas d'apparition d'une neuropathie périphérique de grade ≥ 3 pendant le traitement par BUGVI en association avec la gemcitabine, suspendre le traitement par BUGVI et poursuivre le traitement par gemcitabine à la même dose. Reprendre le traitement par BUGVI à une dose réduite après résolution de la neuropathie périphérique au grade 0 ou 1 (voir rubrique Posologie et mode d'administration). En cas d'apparition d'une neuropathie périphérique de grade ≥ 3 pendant le traitement par BUGVI en association avec le carboplatine, le traitement doit être suspendu jusqu'à la régression de la neuropathie au grade 0 ou 1, et repris en réduisant la dose pour toutes les cures suivantes de BUGVI et de carboplatine (voir rubrique Posologie et mode d'administration).
Sepsis
Un sepsis a été rapporté avec une incidence de 5 % chez les patients présentant ou non une neutropénie qui recevaient le paclitaxel sous forme de nanoparticules liées à l'albumine associé à la gemcitabine. Des complications liées au cancer du pancréas sous-jacent, en particulier une obstruction biliaire ou la présence d'un stent biliaire, ont été identifiées comme étant des facteurs favorisants significatifs. Si un patient développe une fièvre (quel que soit le taux de neutrophiles), instaurer une antibiothérapie à large spectre. En cas de neutropénie fébrile, suspendre l'administration de BUGVI et de gemcitabine jusqu'à ce que la fièvre ait disparu et que le taux de PNN soit ≥ 1 500/mm3, puis reprendre le traitement à des doses réduites (voir rubrique Posologie et mode d'administration).
Pneumopathie inflammatoire
Une pneumopathie inflammatoire est survenue chez 1 % des patients recevant le paclitaxel sous forme de nanoparticules liées à l'albumine en monothérapie et chez 4 % des patients recevant le paclitaxel sous forme de nanoparticules liées à l'albumine en association avec la gemcitabine. Surveiller étroitement tous les patients en vue de détecter tout signe ou symptôme de pneumopathie inflammatoire. Si une pneumopathie inflammatoire est diagnostiquée et qu'une étiologie infectieuse est exclue, arrêter définitivement le traitement par BUGVI et gemcitabine et instaurer rapidement un traitement et des mesures de soutien appropriés (voir rubrique Posologie et mode d'administration).
Insuffisance hépatique
La toxicité du paclitaxel pouvant être accrue en cas d'insuffisance hépatique, l'administration de BUGVI à des patients présentant une insuffisance hépatique doit être effectuée avec prudence. Les patients atteints d'une insuffisance hépatique présentent un risque accru de toxicité, particulièrement en termes de myélosuppression ; ces patients doivent faire l'objet d'une surveillance étroite quant au risque de survenue d'une myélosuppression sévère.
BUGVI n'est pas recommandé chez les patients ayant un taux de bilirubine totale > 5 × LSN ou d'ASAT > 10 × LSN. De plus, BUGVI n'est pas recommandé chez les patients présentant un adénocarcinome du pancréas métastatique atteints d'insuffisance hépatique modérée à sévère (bilirubine totale > 1,5 × LSN et ASAT ≤ 10 × LSN) (voir rubrique Propriétés pharmacocinétiques).
Cardiotoxicité
De rares cas d'insuffisance cardiaque congestive et de dysfonctionnement ventriculaire gauche ont été observés chez des individus recevant le paclitaxel sous forme de nanoparticules liées à l'albumine. La plupart d'entre eux avaient été exposés au préalable à des médicaments cardiotoxiques, tels que les anthracyclines, ou présentaient une pathologie cardiaque sous-jacente. Par conséquent, les patients recevant BUGVI doivent faire l'objet d'une surveillance étroite par leur médecin quant au risque de survenue d'événements cardiaques.
Métastases du SNC
L'efficacité et la sécurité du paclitaxel sous forme de nanoparticules liées à l'albumine chez les patients ayant des métastases au niveau du système nerveux central (SNC) n'ont pas été établies. Les métastases du SNC sont généralement peu contrôlées par la chimiothérapie systémique.
Symptômes gastro-intestinaux
Si les patients présentent des nausées, des vomissements et des diarrhées suite à l'administration de BUGVI, ils peuvent être traités par des agents antiémétiques et antidiarrhéiques fréquemment utilisés.
Affections oculaires
Des cas d'œdème maculaire cystoïde (OMC) ont été rapportés chez des patients traités par paclitaxel sous forme de nanoparticules liées à l'albumine. Les patients présentant des troubles de la vision doivent réaliser rapidement un examen ophtalmologique complet. En cas de diagnostic d'OMC, le traitement par BUGVI doit être arrêté et un traitement approprié instauré (voir rubrique Effets indésirables).
Patients âgés de 75 ans et plus
Chez les patients âgés de 75 ans et plus, aucun bénéfice du traitement par BUGVI en association avec la gemcitabine n'a été démontré par rapport à la gemcitabine en monothérapie. Chez les patients très âgés (≥ 75 ans) ayant reçu le paclitaxel sous forme de nanoparticules liées à l'albumine et la gemcitabine, une incidence plus élevée d'effets indésirables graves et d'effets indésirables ayant entraîné l'arrêt du traitement a été observée, incluant toxicités hématologiques, neuropathie périphérique, appétit diminué et déshydratation.
La capacité à tolérer BUGVI en association avec la gemcitabine doit être évaluée soigneusement chez les patients atteints d'un adénocarcinome du pancréas âgés de 75 ans et plus, en accordant une attention particulière à l'indice de performance, aux comorbidités et au risque accru d'infections (voir rubriques Posologie et mode d'administration et Effets indésirables).
Autres
Bien que les données disponibles soient limitées, aucun bénéfice manifeste n'a été démontré en termes d'allongement de la survie globale chez les patients atteints d'un adénocarcinome du pancréas présentant un taux de CA 19-9 normal avant le début du traitement par paclitaxel sous forme de nanoparticules liées à l'albumine plus gemcitabine (voir rubrique Propriétés pharmacodynamiques).
L'erlotinib ne doit pas être co-administré avec le paclitaxel sous forme de nanoparticules liées à l'albumine plus gemcitabine (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Excipients
Ce médicament contient moins de 1 mmol (23 mg) de sodium par 100 mg, c.-à-d. qu'il est essentiellement « sans sodium ».
Résumé du profil de sécurité
Les effets indésirables cliniquement significatifs les plus fréquents associés à l'utilisation du paclitaxel sous forme de nanoparticules liées à l'albumine ont été une neutropénie, une neuropathie périphérique, des arthralgies/myalgies et des affections gastro-intestinales.
Tableau listant les effets indésirables
Le tableau 6 énumère les effets indésirables associés au paclitaxel sous forme de nanoparticules liées à l'albumine en monothérapie, toutes doses et indications confondues au cours des essais cliniques (N = 789), au paclitaxel en association avec la gemcitabine pour le traitement de l'adénocarcinome pancréatique dans un essai clinique de phase III (N = 421), au paclitaxel en association avec le carboplatine pour le traitement du cancer bronchique non à petites cellules dans un essai clinique de phase III (N = 514) et en utilisation post-commercialisation.
Les fréquences sont définies de la façon suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés selon un ordre décroissant de gravité.
Tableau 6 : Effets indésirables rapportés avec le paclitaxel
|
|
Monothérapie (N = 789) |
Association avec la gemcitabine (N = 421) |
Association avec le carboplatine (N = 514) |
|
Infections et infestations |
|||
|
Fréquent |
Infection, infection des voies urinaires, folliculite, infection des voies aériennes supérieures, candidiase, sinusite. |
Sepsis, pneumonie, candidose orale. |
Pneumonie, bronchite, infection des voies aériennes supérieures, infection des voies urinaires. |
|
Peu fréquent |
Sepsis1, sepsis neutropénique1, pneumonie, candidose orale, rhinopharyngite, cellulite, herpès, infection virale, zona, infection fongique, infection liée au cathéter, infection au site d'injection. |
|
Sepsis, candidose orale. |
|
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes) |
|||
|
Peu fréquent |
Nécrose de la tumeur, douleur métastatique. |
|
|
|
Affections hématologiques et du système lymphatique |
|||
|
Très fréquent |
Aplasie médullaire, neutropénie, thrombopénie, anémie, leucopénie, lymphopénie. |
Neutropénie, thrombopénie, anémie. |
Neutropénie3, thrombopénie3, anémie3, leucopénie3. |
|
Fréquent |
Neutropénie fébrile. |
Pancytopénie. |
Neutropénie fébrile, lymphopénie. |
|
Peu fréquent |
|
Purpura thrombotique thrombocytopénique. |
Pancytopénie. |
|
Rare |
Pancytopénie. |
|
|
|
Affections du système immunitaire |
|||
|
Peu fréquent |
Hypersensibilité. |
|
Hypersensibilité médicamenteuse, hypersensibilité. |
|
Rare |
Hypersensibilité sévère1. |
|
|
|
Troubles du métabolisme et de la nutrition |
|||
|
Très fréquent |
Anorexie. |
Déshydratation, appétit diminué, hypokaliémie. |
Appétit diminué. |
|
Fréquent |
Déshydratation, appétit diminué, hypokaliémie. |
|
Déshydratation. |
|
Peu fréquent |
Hypophosphatémie, rétention hydrique, hypoalbuminémie, polydipsie, hyperglycémie, hypocalcémie, hypoglycémie, hyponatrémie. |
|
|
|
Fréquence indéterminée |
Syndrome de lyse tumorale1. |
|
|
|
Affections psychiatriques |
|||
|
Très fréquent |
|
Dépression, insomnie. |
|
|
Fréquent |
Dépression, insomnie, anxiété. |
Anxiété. |
Insomnie. |
|
Peu fréquent |
Impatiences. |
|
|
|
Affections du système nerveux |
|||
|
Très fréquent |
Neuropathie périphérique, neuropathie, hypoesthésie, paresthésie. |
Neuropathie périphérique, sensations vertigineuses, céphalées, dysgueusie. |
Neuropathie périphérique. |
|
Fréquent |
Neuropathie périphérique sensitive, sensations vertigineuses, neuropathie motrice périphérique, ataxie, céphalées, trouble sensoriel, somnolence, dysgueusie. |
|
Sensations vertigineuses, céphalées, dysgueusie. |
|
Peu fréquent |
Polyneuropathie, aréflexie, syncope, étourdissement postural, dyskinésie, hyporéflexie, névralgie, douleur neuropathique, tremblements, déficit sensoriel. |
Paralysie du septième nerf crânien. |
|
|
Fréquence indéterminée |
Paralysies multiples des nerfs crâniens1. |
|
|
|
Affections oculaires |
|||
|
Fréquent |
Vision trouble, augmentation de la sécrétion lacrymale, sécheresse oculaire, kératoconjonctivite sèche, madarose. |
Augmentation de la sécrétion lacrymale. |
Vision trouble. |
|
Peu fréquent |
Baisse de l'acuité visuelle, vision anormale, irritation oculaire, douleur oculaire, conjonctivite, perturbation visuelle, prurit de l'œil, kératite. |
Œdème maculaire cystoïde. |
|
|
Rare |
Œdème maculaire cystoïde1. |
|
|
|
Affections de l'oreille et du labyrinthe |
|||
|
Fréquent |
Vertige. |
|
|
|
Peu fréquent |
Acouphènes, douleur auriculaire. |
|
|
|
Affections cardiaques |
|||
|
Fréquent |
Arythmies, tachycardie, tachycardie supraventriculaire. |
Insuffisance cardiaque congestive, tachycardie. |
|
|
Rare |
Arrêt cardiaque, insuffisance cardiaque congestive, dysfonctionnement ventriculaire gauche, bloc auriculoventriculaire1, bradycardie. |
|
|
|
Affections vasculaires |
|||
|
Fréquent |
Hypertension, lymphœdème, bouffées congestives, bouffées de chaleur au visage et au cou. |
Hypotension, hypertension. |
Hypotension, hypertension. |
|
Peu fréquent |
Hypotension, hypotension orthostatique, froideur des extrémités. |
Bouffées de chaleur au visage et au cou. |
Bouffées de chaleur au visage et au cou. |
|
Rare |
Thrombose. |
|
|
|
Affections respiratoires, thoraciques et médiastinales |
|||
|
Très fréquent |
|
Dyspnée, épistaxis, toux |
Dyspnée. |
|
Fréquent |
Pneumopathie interstitielle2, dyspnée, épistaxis, douleur pharyngolaryngée, toux, rhinite, rhinorrhée. |
Pneumopathie inflammatoire, congestion nasale. |
Hémoptysie, épistaxis, toux. |
|
Peu fréquent |
Embolie pulmonaire, thromboembolie pulmonaire, épanchement pleural, dyspnée d'exercice, congestion sinusienne, murmure vésiculaire diminué, toux productive, rhinite allergique, enrouement, congestion nasale, sécheresse nasale, sibilances. |
Gorge sèche, sécheresse nasale. |
Pneumopathie inflammatoire. |
|
Fréquence indéterminée |
Parésie des cordes vocales1. |
|
|
|
Affections gastro-intestinales |
|||
|
Très fréquent |
Diarrhée, vomissements, nausées, constipation, stomatite. |
Diarrhée, vomissements, nausées, constipation, douleur abdominale, douleur abdominale haute. |
Diarrhée, vomissements, nausées, constipation. |
|
Fréquent |
Reflux gastro-œsophagien, dyspepsie, douleur abdominale, distension abdominale, douleur abdominale haute, hypoesthésie orale. |
Occlusion intestinale, colite, stomatite, bouche sèche. |
Stomatite, dyspepsie, dysphagie, douleur abdominale. |
|
Peu fréquent |
Hémorragie rectale, dysphagie, flatulences, glossodynie, bouche sèche, douleur gingivale, selles trop liquides, œsophagite, douleur abdominale basse, ulcération buccale, douleur buccale. |
|
|
|
Affections hépatobiliaires |
|||
|
Fréquent |
|
Cholangite. |
Hyperbilirubinémie. |
|
Peu fréquent |
Hepatomégalie. |
|
|
|
Affections de la peau et du tissu sous-cutané |
|||
|
Très fréquent |
Alopécie, rash. |
Alopécie, rash. |
Alopécie, rash. |
|
Fréquent |
Prurit, sécheresse cutanée, trouble unguéal, érythème, altération de la couleur unguéale/pigmentation unguéale, hyperpigmentation cutanée, onycholyse, changements unguéaux. |
Prurit, sécheresse cutanée, trouble unguéal. |
Prurit, trouble unguéal. |
|
Peu fréquent |
Réaction de photosensibilité, urticaire, peau douloureuse, prurit généralisé, éruption prurigineuse, trouble de la peau, trouble pigmentaire, hyperhidrose, onychomadèse, rash érythémateux, rash généralisé, dermatite, sueurs nocturnes, rash maculopapuleux, vitiligo, hypotrichose, sensibilité du lit de l'ongle, gêne unguéale, rash maculeux, rash papuleux, lésion de la peau, œdème du visage. |
|
Exfoliation cutanée, dermatite allergique, urticaire. |
|
Très rare |
Syndrome de Stevens-Johnson1, nécrolyse épidermique toxique1. |
|
|
|
Fréquence indéterminée |
Syndrome d'érythrodysesthésie palmo-plantaire1,4, sclérodermie1. |
|
|
|
Affections musculosquelettiques et du tissu conjonctif |
|||
|
Très fréquent |
Arthralgie, myalgie. |
Arthralgie, myalgie, douleur des extrémités. |
Arthralgie, myalgie. |
|
Fréquent |
Dorsalgie, douleur des extrémités, douleur osseuse, crampes musculaires, douleur des membres. |
Faiblesse musculaire, douleur osseuse. |
Dorsalgie, douleur des extrémités, douleur musculosquelettique. |
|
Peu fréquent |
Douleur de la paroi thoracique, faiblesse musculaire, douleur dans le cou, douleur de l'aine, spasmes musculaires, douleur musculosquelettique, douleur du flanc, gêne dans les membres, faiblesse musculaire. |
|
|
|
Affections du rein et des voies urinaires |
|||
|
Fréquent |
|
Insuffisance rénale aiguë. |
|
|
Peu fréquent |
Hématurie, dysurie, pollakiurie, nycturie, polyurie, incontinence urinaire. |
Syndrome hémolytique et urémique. |
|
|
Affections des organes de reproduction et du sein |
|||
|
Peu fréquent |
Douleur mammaire. |
|
|
|
Troubles généraux et anomalies au site d'administration |
|||
|
Très fréquent |
Fatigue, asthénie, fièvre. |
Fatigue, asthénie, fièvre, œdème périphérique, frissons. |
Fatigue, asthénie, œdème périphérique. |
|
Fréquent |
Malaise, léthargie, faiblesse, œdème périphérique, inflammation muqueuse, douleur, frissons, œdème, diminution de l'indice de performance, douleur thoracique, syndrome grippal, hyperpyrexie. |
Réaction au site de perfusion. |
Fièvre, douleur thoracique. |
|
Peu fréquent |
Gêne thoracique, démarche anormale, gonflement, réaction au site d'injection. |
|
Inflammation muqueuse, extravasation au site de perfusion, inflammation au site de perfusion, rash au site de perfusion. |
|
Rare |
Extravasation. |
|
|
|
Investigations |
|||
|
Très fréquent |
|
Poids diminué, alanine aminotransférase augmentée. |
|
|
Fréquent |
Poids diminué, alanine aminotransférase augmentée, aspartate aminotransférase augmentée, hématocrite diminué, globules rouges diminués, température augmentée, gamma-glutamyltransférase augmentée, phosphatase alcaline sanguine augmentée. |
Aspartate aminotransférase augmentée, bilirubine sanguine augmentée, créatine sanguine augmentée. |
Poids diminué, alanine aminotransférase augmentée, aspartate aminotransférase augmentée, phosphatase alcaline sanguine augmentée. |
|
Peu fréquent |
Pression artérielle augmentée, poids augmenté, lactate déshydrogénase sanguine augmentée, créatinine sanguine augmentée, glucose sanguin augmenté, phosphore sanguin augmenté, potassium sanguin diminué, bilirubine augmentée. |
|
|
|
Lésions, intoxications et complications d'interventions |
|||
|
Peu fréquent |
Contusion. |
|
|
|
Rare |
Réaction cutanée par réactivation de la zone antérieurement irradiée, pneumopathie radique. |
|
|
1 Rapportés dans le cadre de la surveillance post-commercialisation du paclitaxel sous forme de nanoparticules liées à l'albumine.
2 La fréquence des pneumopathies inflammatoires est calculée à partir des données combinées de 1 310 patients inclus dans les essais cliniques recevant le paclitaxel sous forme de nanoparticules liées à l'albumine en monothérapie pour un cancer du sein et dans d'autres indications.
3 Sur la base des analyses biologiques : degré maximal de myélosuppression (population traitée).
4 Chez certains patients ayant été exposés à la capécitabine.
Description de certains effets indésirables
Cette rubrique décrit les effets indésirables les plus fréquents et les plus cliniquement pertinents associés au paclitaxel sous forme de nanoparticules liées à l'albumine.
Les effets indésirables ont été évalués chez 229 patients présentant un cancer du sein métastatique ayant été traités par paclitaxel sous forme de nanoparticules liées à l'albumine à la dose de 260 mg/m2 toutes les trois semaines au cours de l'étude clinique pivot de phase III (paclitaxel sous forme de nanoparticules liées à l'albumine en monothérapie).
Les effets indésirables ont été évalués chez 421 patients présentant un cancer du pancréas métastatique ayant été traités par paclitaxel sous forme de nanoparticules liées à l'albumine en association avec la gemcitabine (paclitaxel sous forme de nanoparticules liées à l'albumine à la dose de 125 mg/m2 en association avec la gemcitabine à la dose de 1 000 mg/m2, administrée les Jours 1, 8 et 15 de chaque cycle de 28 jours) et 402 patients recevant la gemcitabine en monothérapie en traitement systémique de première ligne d'un adénocarcinome du pancréas métastatique (paclitaxel/gemcitabine).
Les effets indésirables ont été évalués chez 514 patients atteints d'un cancer bronchique non à petites cellules ayant été traités par paclitaxel sous forme de nanoparticules liées à l'albumine en association avec le carboplatine (100 mg/m2 de paclitaxel, administré les Jours 1, 8 et 15 de chaque cycle de 21 jours en association avec le carboplatine administré le Jour 1 de chaque cycle) dans un essai clinique randomisé, contrôlé de phase III (paclitaxel sous forme de nanoparticules liées à l'albumine/carboplatine). La toxicité des taxanes rapportée par les patients a été évaluée à l'aide des 4 sous-échelles du questionnaire d'évaluation fonctionnelle du traitement anticancéreux Functional Assessment of Cancer Therapy(FACT)-Taxane. Dans une analyse pour mesures répétées, 3 des 4 sous-échelles (neuropathie périphérique, douleurs dans les mains/pieds et audition) ont été en faveur du paclitaxel sous forme de nanoparticules liées à l'albumine en association avec le carboplatine (p ≤ 0,002). Pour l'autre sous-échelle (œdème), aucune différence n'a été observée entre les bras de traitement.
Infections et infestations
Paclitaxel sous forme de nanoparticules liées à l'albumine/gemcitabine
Un sepsis a été rapporté à un taux de 5 % chez les patients présentant ou non une neutropénie, qui recevaient le paclitaxel sous forme de nanoparticules liées à l'albumine en association avec la gemcitabine au cours d'un essai mené dans l'adénocarcinome du pancréas. Sur les 22 cas de sepsis rapportés chez les patients traités par paclitaxel sous forme de nanoparticules liées à l'albumine en association avec la gemcitabine, 5 ont été d'issue fatale. Des complications liées au cancer du pancréas sous-jacent, en particulier une obstruction biliaire ou la présence d'un stent biliaire, ont été identifiés comme étant des facteurs favorisants significatifs. Si un patient développe une fièvre (quel que soit le taux de neutrophiles), instaurer une antibiothérapie à large spectre. En cas de neutropénie fébrile, suspendre l'administration de BUGVI et de gemcitabine jusqu'à ce que la fièvre ait disparu et que le taux de PNN soit ≥ 1 500/mm3, puis reprendre le traitement à des doses réduites (voir rubrique Posologie et mode d'administration).
Affections hématologiques et du système lymphatique
Paclitaxel sous forme de nanoparticules liées à l'albumine en monothérapie — cancer du sein métastatique
Chez les patients atteints d'un cancer du sein métastatique, la neutropénie se distinguait comme la plus importante toxicité hématologique (rapportée chez 79 % des patients) et était rapidement réversible et dose-dépendante ; la leucopénie a été rapportée chez 71 % des patients. Une neutropénie de grade 4 (< 500 cellules/mm3) est survenue chez 9 % des patients traités par paclitaxel sous forme de nanoparticules liées à l'albumine. Une neutropénie fébrile est survenue chez quatre patients sous paclitaxel sous forme de nanoparticules liées à l'albumine. Une anémie (Hb < 10 g/dL) a été observée chez 46 % des patients sous paclitaxel sous forme de nanoparticules liées à l'albumine et a été sévère (Hb < 8 g/dL) dans trois cas.
Une lymphopénie a été observée chez 45 % des patients.
Paclitaxel sous forme de nanoparticules liées à l'albumine/gemcitabine
Le tableau 7 présente la fréquence et la sévérité des anomalies des paramètres hématologiques chez les patients traités par paclitaxel sous forme de nanoparticules liées à l'albumine en association avec la gemcitabine ou par gemcitabine en monothérapie.
Tableau 7 : Anomalies des paramètres hématologiques dans l'essai sur l'adénocarcinome du pancréas
|
|
Paclitaxel sous forme de nanoparticules liées à l'albumine (125 mg/m2)/gemcitabine |
Gemcitabine |
||
|
Grades 1-4 (%) |
Grades 3-4 (%) |
Grades 1-4 (%) |
Grades 3-4 (%) |
|
|
Anémiea,b |
97 |
13 |
96 |
12 |
|
Neutropéniea,b |
73 |
38 |
58 |
27 |
|
Thrombopéniea,b |
74 |
13 |
70 |
9 |
a 405 patients évalués dans le groupe traité par paclitaxel sous forme de nanoparticules liées à l'albumine/gemcitabine.
b 388 patients évalués dans le groupe traité par gemcitabine.
c 404 patients évalués dans le groupe traité par paclitaxel sous forme de nanoparticules liées à l'albumine/gemcitabine.
Paclitaxel sous forme de nanoparticules liées à l'albumine/carboplatine
L'anémie et la thrombopénie ont été rapportées plus fréquemment dans le bras paclitaxel sous forme de nanoparticules liées à l'albumine et carboplatine que dans le bras paclitaxel et carboplatine (54 % versus 28 % et 45 % versus 27 %, respectivement).
Affections du système nerveux
Paclitaxel sous forme de nanoparticules liées à l'albumine en monothérapie — cancer du sein métastatique
Généralement, la fréquence et la sévérité de la neurotoxicité étaient dose-dépendantes chez les patients recevant le paclitaxel sous forme de nanoparticules liées à l'albumine. Des neuropathies périphériques (le plus souvent des neuropathies sensitives de grade 1 ou 2) ont été observées chez 68 % des patients sous paclitaxel sous forme de nanoparticules liées à l'albumine dont 10 % de grade 3, et aucune de grade 4.
Paclitaxel sous forme de nanoparticules liées à l'albumine/gemcitabine
Chez les patients traités par paclitaxel sous forme de nanoparticules liées à l'albumine en association avec la gemcitabine, le délai médian jusqu'à la première survenue d'une neuropathie périphérique de grade 3 a été de 140 jours. Le délai médian jusqu'à l'amélioration d'au moins un grade a été de 21 jours et le délai médian jusqu'à la régression de la neuropathie périphérique du grade 3 au grade 0 ou 1 a été de 29 jours. Chez les patients dont le traitement avait été interrompu en raison d'une neuropathie périphérique, le traitement par paclitaxel a pu être repris à une dose réduite pour 44 % d'entre eux (31 patients sur 70). Aucun patient traité par paclitaxel sous forme de nanoparticules liées à l'albumine en association avec la gemcitabine n'a présenté de neuropathie périphérique de grade 4.
Paclitaxel sous forme de nanoparticules liées à l'albumine/carboplatine
Chez les patients atteints d'un cancer bronchique non à petites cellules traités par paclitaxel sous forme de nanoparticules liées à l'albumine et le carboplatine, le délai médian jusqu'à la première survenue d'une neuropathie périphérique de grade 3 liée au traitement a été de 121 jours et le délai médian jusqu'à la régression du grade 3 au grade 1 a été de 38 jours. Aucun patient traité par paclitaxel sous forme de nanoparticules liées à l'albumine et carboplatine n'a présenté de neuropathie périphérique de grade 4.
Affections oculaires
Au cours de la surveillance post-commercialisation, de rares cas de baisse d'acuité visuelle due à un œdème maculaire cystoïde ont été rapportés pendant le traitement par paclitaxel sous forme de nanoparticules liées à l'albumine (voir rubrique Mises en garde spéciales et précautions d'emploi).
Affections respiratoires, thoraciques et médiastinales
Paclitaxel sous forme de nanoparticules liées à l'albumine/gemcitabine
Une pneumopathie inflammatoire a été rapportée à un taux de 4 % avec l'utilisation du paclitaxel sous forme de nanoparticules liées à l'albumine en association avec la gemcitabine. Sur les 17 cas de pneumopathie inflammatoire rapportés chez les patients traités par paclitaxel sous forme de nanoparticules liées à l'albumine en association avec la gemcitabine, 2 ont été d'issue fatale. Les patients doivent faire l'objet d'une surveillance étroite en vue de détecter tout signe ou symptôme de pneumopathie inflammatoire. Si une pneumopathie inflammatoire est diagnostiquée et qu'une étiologie infectieuse est exclue, arrêter définitivement le traitement par BUGVI et gemcitabine et instaurer rapidement un traitement approprié et des mesures de soutien (voir rubrique Posologie et mode d'administration).
Affections gastro-intestinales
Paclitaxel sous forme de nanoparticules liées à l'albumine en monothérapie - cancer du sein métastatique
Des nausées ont été rapportées chez 29 % des patients et une diarrhée a été rapportée chez 25 % des patients.
Affections de la peau et du tissu sous-cutané
Paclitaxel sous forme de nanoparticules liées à l'albumine en monothérapie - cancer du sein métastatique
Une alopécie a été observée chez > 80 % des patients traités par paclitaxel sous forme de nanoparticules liées à l'albumine. Dans la majorité des cas, l'alopécie est survenue moins d'un mois après l'instauration du traitement par paclitaxel sous forme de nanoparticules liées à l'albumine. Une perte des cheveux importante de ≥ 50 % est attendue chez la majorité des patients présentant une alopécie.
Affections musculosquelettiques et du tissu conjonctif
Paclitaxel sous forme de nanoparticules liées à l'albumine en monothérapie - cancer du sein métastatique
Des arthralgies sont survenues chez 32 % des patients sous paclitaxel sous forme de nanoparticules liées à l'albumine et ont été sévères dans 6 % des cas. Des myalgies sont survenues chez 24 % des patients sous paclitaxel sous forme de nanoparticules liées à l'albumine et ont été sévères dans 7 % des cas. Les symptômes étaient habituellement transitoires, apparaissaient généralement trois jours après l'administration du paclitaxel sous forme de nanoparticules liées à l'albumine et étaient résolus dans la semaine suivante.
Troubles généraux et anomalies au site d'administration
Paclitaxel sous forme de nanoparticules liées à l'albumine en monothérapie - cancer du sein métastatique
Une asthénie/fatigue a été rapportée chez 40 % des patients.
Population pédiatrique
L'étude a comptabilisé 106 patients, parmi lesquels 104 étaient des patients pédiatriques âgés de 6 mois à moins de 18 ans (voir rubrique Propriétés pharmacodynamiques). Chaque patient a présenté au moins 1 effet indésirable. Les effets indésirables les plus fréquemment rapportés ont été la neutropénie, l'anémie, la leucopénie et la fièvre. Les effets indésirables graves rapportés chez plus de 2 patients ont été la fièvre, les dorsalgies, l'œdème périphérique et les vomissements. Aucun nouveau signal relatif à la sécurité d'emploi n'a été identifié parmi le nombre limité de patients pédiatriques traités par paclitaxel sous forme de nanoparticules liées à l'albumine, et le profil de sécurité a été similaire à celui observé dans la population adulte.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament.
Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : https://signalement.social-sante.gouv.fr/.
Ce médicament est une
formulation de nanoparticules de paclitaxel-albumine pouvant avoir des
propriétés pharmacologiques très différentes des autres formulations de
paclitaxel. NE PAS LE REMPLACER par d'autres formulations de paclitaxel ou
l'utiliser à leur place.
SURVEILLANCE du traitement :
- NFS tout au long du traitement,
- cardiaque,
- symptômes de pneumopathie inflammatoire.
PATIENTS DE SEXE MASCULIN :
- RECOMMANDER de se renseigner sur les procédures de conservation du sperme en raison du risque potentiel de stérilité irréversible lié au traitement.
- UTILISER une méthode de contraception efficace et éviter de concevoir pendant la durée du traitement et jusqu'à six mois après son arrêt.
FEMMES EN AGE DE PROCREER :
- REALISER un test de grossesse avant le début du traitement.
- UTILISER une contraception efficace pendant le traitement et jusqu'à un mois après son arrêt.
SURVEILLANCE du traitement :
- NFS tout au long du traitement,
- cardiaque,
- symptômes de pneumopathie inflammatoire.
PATIENTS DE SEXE MASCULIN :
- RECOMMANDER de se renseigner sur les procédures de conservation du sperme en raison du risque potentiel de stérilité irréversible lié au traitement.
- UTILISER une méthode de contraception efficace et éviter de concevoir pendant la durée du traitement et jusqu'à six mois après son arrêt.
FEMMES EN AGE DE PROCREER :
- REALISER un test de grossesse avant le début du traitement.
- UTILISER une contraception efficace pendant le traitement et jusqu'à un mois après son arrêt.
Contraception chez les hommes et les femmes
Les femmes en capacité de procréer doivent utiliser une méthode de contraception efficace pendant le traitement par paclitaxel et jusqu'à 1 mois après l'arrêt du traitement. Il est conseillé aux hommes traités par paclitaxel d'utiliser une méthode de contraception efficace et d'éviter de concevoir un enfant pendant toute la durée du traitement et jusqu'à six mois après l'arrêt du traitement.
Grossesse
Il existe des données très limitées sur l'utilisation du paclitaxel chez la femme enceinte. Le paclitaxel est susceptible de provoquer des malformations graves s'il est administré pendant la grossesse. Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique Données de sécurité préclinique). Les femmes en capacité de procréer doivent réaliser un test de grossesse avant de débuter le traitement par paclitaxel.
Le paclitaxel ne doit pas être utilisé pendant la grossesse ni chez la femme en capacité de procréer n'utilisant pas de méthode de contraception efficace, sauf si l'état clinique de la mère nécessite un traitement par paclitaxel.
Allaitement
Le paclitaxel et/ou ses métabolites sont excrétés dans le lait de rates allaitantes (voir rubrique Données de sécurité préclinique). On ne sait pas si le paclitaxel est excrété dans le lait maternel. Compte tenu des effets indésirables graves potentiels pour les nouveau-nés allaités, le paclitaxel est contre-indiqué pendant l'allaitement. L'allaitement doit être interrompu pendant la durée du traitement.
Fertilité
Le paclitaxel sous forme de nanoparticules liées à l'albumine a induit une infertilité chez les rats mâles (voir rubrique Données de sécurité préclinique). Selon les résultats obtenus sur les animaux, la fertilité des hommes et des femmes peut être compromise. Avant de débuter le traitement, il est conseillé aux patients de sexe masculin de se renseigner sur les procédures de conservation du sperme en raison du risque potentiel d'infertilité irréversible due au traitement par paclitaxel.
Le paclitaxel est partiellement métabolisé par les isoenzymes CYP2C8 et CYP3A4 du cytochrome P450 (voir rubrique Propriétés pharmacocinétiques). Par conséquent, en l'absence d'étude pharmacocinétique d'interactions médicamenteuses, la prudence est de mise lors de l'administration de paclitaxel en association avec des médicaments inhibiteurs du CYP2C8 ou du CYP3A4 (par exemple, kétoconazole et autres antifongiques imidazolés, érythromycine, fluoxétine, gemfibrozil, clopidogrel, cimétidine, ritonavir, saquinavir, indinavir et nelfinavir), car la toxicité du paclitaxel peut être accrue compte tenu de l'augmentation de l'exposition au paclitaxel. L'administration de paclitaxel en association avec des médicaments inducteurs du CYP2C8 ou du CYP3A4 (rifampicine, carbamazépine, phénytoïne, éfavirenz, névirapine) n'est pas recommandée, car l'efficacité peut être compromise compte tenu de la diminution de l'exposition au paclitaxel.
Le paclitaxel et la gemcitabine n'ont pas de voie métabolique commune. La clairance du paclitaxel est due essentiellement au métabolisme médié par les CYP2C8 et CYP3A4, suivi d'une excrétion biliaire, tandis que la gemcitabine est inactivée par la cytidine désaminase, suivie d'une excrétion urinaire. Les interactions pharmacocinétiques entre le paclitaxel sous forme de nanoparticules liées à l'albumine et la gemcitabine n'ont pas été évaluées chez l'être humain.
Une étude pharmacocinétique a été menée avec le paclitaxel sous forme de nanoparticules liées à l'albumine et le carboplatine chez des patients présentant un cancer bronchique non à petites cellules. Aucune interaction pharmacocinétique cliniquement pertinente n'a été observée entre le paclitaxel sous forme de nanoparticules liées à l'albumine et le carboplatine.
BUGVI est indiqué en monothérapie dans le traitement du cancer du sein, en association avec la gemcitabine dans le traitement de l'adénocarcinome du pancréas ou en association avec le carboplatine dans le traitement du cancer bronchique non à petites cellules (voir rubrique Indications thérapeutiques). BUGVI ne doit pas être utilisé en association avec d'autres agents anticancéreux.
Population pédiatrique
Les études d'interaction n'ont été réalisées que chez l'adulte.
BUGVI ne doit être administré que sous la responsabilité d'un oncologue qualifié au sein d'une unité spécialisée dans l'administration d'agents cytotoxiques. Il ne doit ni remplacer ni être substitué par d'autres formulations de paclitaxel.
Posologie
Cancer du sein
La dose recommandée de BUGVI est de 260 mg/m2 administrée en perfusion intraveineuse de 30 minutes toutes les 3 semaines.
Adaptations posologiques au cours du traitement du cancer du sein
Les patients présentant une neutropénie sévère (taux de neutrophiles < 500/mm3 pendant au moins une semaine) ou une neuropathie sensitive sévère au cours du traitement par BUGVI doivent recevoir une dose réduite à 220 mg/m2 pour les cures suivantes. Suite à la récidive d'une neutropénie sévère ou d'une neuropathie sensitive sévère, la dose doit être à nouveau réduite à 180 mg/m2. BUGVI ne doit pas être administré avant que le taux de neutrophiles ne redevienne > 1 500/mm3. Pour les neuropathies sensitives de grade 3, suspendre le traitement jusqu'à ce que la neuropathie régresse au grade 1 ou 2, et réduire la dose administrée pour l'ensemble des cures suivantes.
Adénocarcinome du pancréas
La dose recommandée de BUGVI en association avec la gemcitabine est de 125 mg/m2 administrée en perfusion intraveineuse de 30 minutes les Jours 1, 8 et 15 de chaque cycle de 28 jours. La dose concomitante recommandée de gemcitabine est de 1 000 mg/m2 administrée en perfusion intraveineuse de 30 minutes immédiatement après la fin de l'administration de BUGVI les Jours 1, 8 et 15 de chaque cycle de 28 jours.
Adaptations posologiques au cours du traitement de l'adénocarcinome du pancréas
Tableau 1 : Réductions des paliers de dose chez les patients présentant un adénocarcinome du pancréas
|
Palier de dose |
Dose de BUGVI (mg/m2) |
Dose de gemcitabine (mg/m2) |
|
Dose complète |
125 |
1 000 |
|
1re réduction de dose |
100 |
800 |
|
2e réduction de dose |
75 |
600 |
|
Si une réduction supplémentaire de la dose est nécessaire |
Arrêter le traitement |
Arrêter le traitement |
Tableau 2 : Modifications de doses en cas de neutropénie et/ou de thrombopénie au début d'un cycle ou pendant un cycle chez les patients présentant un adénocarcinome du pancréas
|
Jour du cycle |
Taux de PNN (cellules/mm3) |
|
Taux de plaquettes (cellules/mm3) |
Dose de BUGVI |
Dose de gemcitabine |
|
Jour 1 |
< 1 500 |
OU |
< 100 000 |
Différer le traitement jusqu'à la récupération |
|
|
Jour 8 |
≥ 500 mais < 1 000 |
OU |
≥ 50 000 mais < 75 000 |
Réduire les doses d'un palier |
|
|
|
< 500 |
OU |
< 50 000 |
Interrompre le traitement |
|
|
Jour 15 : Si les doses du Jour 8 ont été administrées sans modification : |
|||||
|
Jour 15 |
≥ 500 mais < 1 000 |
OU |
≥ 50 000 mais < 75 000 |
Traiter au palier de dose du Jour 8 puis administrer des facteurs de croissance leucocytaire OU Réduire les doses du Jour 8 d'un palier |
|
|
|
< 500 |
OU |
< 50 000 |
Interrompre le traitement |
|
|
Jour 15 : Si les doses du Jour 8 ont été réduites : |
|||||
|
Jour 15 |
≥ 1 000 |
ET |
≥ 75 000 |
Revenir aux paliers de dose du Jour 1 puis administrer des facteurs de croissance leucocytaire OU Traiter aux doses du Jour 8 |
|
|
|
≥ 500 mais < 1 000 |
OU |
≥ 50 000 mais < 75 000 |
Traiter aux paliers de dose du Jour 8 puis administrer des facteurs de croissance leucocytaire OU Réduire les doses du Jour 8 d'un palier |
|
|
|
< 500 |
OU |
< 50 000 |
Interrompre le traitement |
|
|
Jour 15 : Si l'administration du traitement a été suspendue le Jour 8 : |
|||||
|
Jour 15 |
≥ 1 000 |
ET |
≥ 75 000 |
Revenir aux paliers de dose du Jour 1 puis administrer des facteurs de croissance leucocytaire OU Réduire les doses du Jour 1 d'un palier |
|
|
|
≥ 500 mais < 1 000 |
OU |
≥ 50 000 mais < 75 000 |
Réduire d'un palier de dose puis administrer des facteurs de croissance leucocytaire OU Réduire les doses du Jour 1 de deux paliers |
|
|
|
< 500 |
OU |
< 50 000 |
Interrompre le traitement |
|
Abréviation : PNN = polynucléaires neutrophiles
Tableau 3 : Modifications de doses en cas de survenue d'autres effets indésirables chez les patients présentant un adénocarcinome du pancréas
|
Effet indésirable (EI) |
Dose de BUGVI |
Dose de gemcitabine |
|
Neutropénie fébrile : Grade 3 ou 4 |
Interrompre le traitement jusqu'à résolution de la fièvre et taux de PNN ≥ 1 500 ; reprendre le traitement au palier de dose immédiatement inférieura |
|
|
Neuropathie périphérique : Grade 3 ou 4 |
Interrompre le traitement jusqu'à régression au grade ≤ 1 ; reprendre le traitement au palier de dose immédiatement inférieura |
Traiter à la même dose |
|
Toxicité cutanée : Grade 2 ou 3 |
Diminuer au palier de dose immédiatement inférieura ; si l'EI persiste, arrêter le traitement |
|
|
Toxicité gastro-intestinale : Mucite ou diarrhée de grade 3 |
Interrompre le traitement jusqu'à régression au grade ≤ 1 ; reprendre le traitement au palier de dose immédiatement inférieura |
|
a Voir le tableau 1 pour les réductions de doses
Cancer bronchique non à petites cellules
La dose recommandée de BUGVI est de 100 mg/m2 administrée en perfusion intraveineuse de 30 minutes les Jours 1, 8 et 15 de chaque cycle de 21 jours. La dose recommandée de carboplatine est ASC = 6 mg•min/mL, uniquement le Jour 1 de chaque cycle de 21 jours, immédiatement après la fin de l'administration de BUGVI.
Adaptations posologiques au cours du traitement du cancer bronchique non à petites cellules
BUGVI ne doit être administré le Jour 1 d'un cycle que si le taux de polynucléaires neutrophiles (PNN) est ≥ 1 500/mm3 et le taux de plaquettes ≥ 100 000/mm3. Pour chaque administration hebdomadaire ultérieure de BUGVI, les patients doivent avoir un taux de PNN ≥ 500/mm3 et un taux de plaquettes > 50 000/mm3 sinon le traitement doit être suspendu jusqu'à la récupération. Après récupération des taux, reprendre le traitement la semaine suivante conformément aux critères présentés dans le tableau 4. La dose suivante ne doit être réduite que si les critères présentés dans le tableau 4 sont remplis.
Tableau 4 : Réductions de dose en cas de toxicités hématologiques chez les patients présentant un cancer bronchique non à petites cellules
|
Toxicité hématologique |
Survenue |
Dose de BUGVI (mg/m2)1 |
Dose de carboplatine (ASC mg•min/mL)1 |
|
Nadir des PNN < 500/mm3 avec fièvre neutropénique > 38 °C OU Report du prochain cycle en raison d'une neutropénie persistante2 (nadir des PNN < 1 500/mm3) OU Nadir des PNN < 500/mm3 pendant > 1 semaine |
Première |
75 |
4,5 |
|
Deuxième |
50 |
3,0 |
|
|
Troisième |
Arrêter le traitement |
||
|
Nadir des plaquettes < 50 000/mm3 |
Première |
75 |
4,5 |
|
Deuxième |
Arrêter le traitement |
||
1 Le Jour 1 du cycle de 21 jours, réduire simultanément les doses de BUGVI et de carboplatine. Les Jours 8 ou 15 du cycle de 21 jours, réduire la dose de BUGVI ; réduire la dose de carboplatine lors du cycle suivant.
2 Pendant 7 jours maximum après la dose programmée du Jour 1 du prochain cycle.
En cas de toxicité cutanée de grade 2 ou 3, de diarrhée de grade 3 ou de mucite de grade 3, interrompre le traitement jusqu'à la régression de la toxicité à un grade ≤ 1, puis reprendre le traitement conformément aux recommandations figurant dans le tableau 5. En cas de neuropathie périphérique de grade ≥ 3, interrompre le traitement jusqu'à la régression de la neuropathie à un grade ≤ 1. Le traitement pourra être repris au palier de dose immédiatement inférieur lors des cycles suivants, conformément aux recommandations figurant dans le tableau 5. Pour toute autre toxicité non hématologique de grade 3 ou 4, interrompre le traitement jusqu'à la régression de la toxicité à un grade ≤ 2, puis reprendre le traitement conformément aux recommandations figurant dans le tableau 5.
Tableau 5 : Réductions de dose en cas de toxicités non hématologiques chez les patients présentant un cancer bronchique non à petites cellules
|
Toxicité non hématologique |
Survenue |
Dose de BUGVI (mg/m2)1 |
Dose de carboplatine (ASC mg•min/mL)1 |
|
Toxicité cutanée de grade 2 ou 3 Diarrhée de grade 3 Mucite de grade 3 Neuropathie périphérique de grade ≥ 3 Toute autre toxicité non hématologique de grade 3 ou 4 |
Première |
75 |
4,5 |
|
Deuxième |
50 |
3,0 |
|
|
Troisième |
Arrêter le traitement |
||
|
Toxicité cutanée, diarrhée ou mucite de grade 4 |
Première |
Arrêter le traitement |
|
1 Le Jour 1 du cycle de 21 jours, réduire simultanément les doses de BUGVI et de carboplatine. Les Jours 8 ou 15 du cycle de 21 jours, réduire la dose de BUGVI ; réduire la dose de carboplatine lors du cycle suivant.
Populations particulières
Insuffisance hépatique
Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère (bilirubine totale > 1 et ≤ 1,5 x LSN et aspartate aminotransférase [ASAT] ≤ 10 × LSN), quelle que soit l'indication. Ces patients doivent être traités aux mêmes doses que ceux présentant une fonction hépatique normale.
Chez les patients présentant un cancer du sein métastatique ou un cancer bronchique non à petites cellules atteints d'insuffisance hépatique modérée à sévère (bilirubine totale > 1,5 et ≤ 5 × LSN et ASAT ≤ 10 × LSN), une réduction de 20 % de la dose est recommandée. La dose réduite pourra être augmentée à la dose utilisée chez les patients présentant une fonction hépatique normale si le patient tolère le traitement pendant au moins deux cycles (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacocinétiques).
Chez les patients présentant un adénocarcinome du pancréas métastatique atteints d'insuffisance hépatique modérée à sévère, les données sont insuffisantes pour établir des recommandations posologiques (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacocinétiques).
Chez les patients présentant un taux de bilirubine totale > 5 × LSN ou d'ASAT > 10 × LSN, les données sont insuffisantes pour établir des recommandations posologiques, quelle que soit l'indication (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacocinétiques).
Insuffisance rénale
Il n'est pas nécessaire d'adapter la dose initiale de BUGVI chez les patients présentant une insuffisance rénale légère à modérée (clairance de la créatinine estimée ≥ 30 à < 90 mL/min). Il n'existe pas de données suffisantes pour recommander une adaptation de la dose de BUGVI chez les patients présentant une insuffisance rénale sévère ou terminale (clairance de la créatinine estimée < 30 mL/min) (voir rubrique Propriétés pharmacocinétiques).
Personnes âgées
Aucune réduction de dose supplémentaire, autre que les réductions indiquées pour tous les patients, n'est recommandée chez les patients âgés de 65 ans et plus.
Chez les 229 patients ayant reçu le paclitaxel sous forme de nanoparticules liées à l'albumine en monothérapie dans le traitement d'un cancer du sein dans l'étude randomisée, 13 % étaient âgés d'au moins 65 ans et < 2 % étaient âgés de 75 ans et plus. Aucune toxicité n'a été observée plus fréquemment chez les patients âgés d'au moins 65 ans ayant reçu le paclitaxel sous forme de nanoparticules liées à l'albumine. Cependant, une analyse ultérieure portant sur 981 patients recevant le paclitaxel sous forme de nanoparticules liées à l'albumine en monothérapie pour un cancer du sein métastatique, parmi lesquels 15 % étaient âgés de ≥ 65 ans et 2 % de ≥ 75 ans, a révélé une incidence plus élevée d'épistaxis, de diarrhée, de déshydratation, de fatigue et d'œdème périphérique chez les patients âgés de ≥ 65 ans.
Chez les 421 patients atteints d'un adénocarcinome du pancréas ayant reçu le paclitaxel sous forme de nanoparticules liées à l'albumine en association avec la gemcitabine dans l'étude randomisée, 41 % étaient âgés de 65 ans et plus et 10 % étaient âgés de 75 ans et plus. Chez les patients âgés de 75 ans et plus ayant reçu le paclitaxel sous forme de nanoparticules liées à l'albumine et la gemcitabine, une incidence plus élevée d'effets indésirables graves et d'effets indésirables ayant entraîné l'arrêt du traitement ont été observés (voir rubrique Mises en garde spéciales et précautions d'emploi). Les patients atteints d'un adénocarcinome du pancréas âgés de 75 ans et plus doivent faire l'objet d'une évaluation attentive avant d'envisager le traitement (voir rubrique Mises en garde spéciales et précautions d'emploi).
Chez les 514 patients présentant un cancer bronchique non à petites cellules ayant reçu le paclitaxel sous forme de nanoparticules liées à l'albumine en association avec le carboplatine dans l'étude randomisée, 31 % étaient âgés de 65 ans et plus et 3,5 % étaient âgés de 75 ans et plus. Les événements de myélosuppression, de neuropathie périphérique et d'arthralgie ont été plus fréquents chez les patients âgés de 65 ans et plus que chez les patients de moins de 65 ans. Les données sur l'utilisation du paclitaxel sous forme de nanoparticules liées à l'albumine en association avec le carboplatine chez les patients âgés de 75 ans et plus sont limitées.
Une modélisation pharmacocinétique/pharmacodynamique utilisant les données de 125 patients présentant des tumeurs solides avancées indiquent que les patients âgés de ≥ 65 ans peuvent être plus susceptibles de développer une neutropénie pendant le premier cycle de traitement.
Population pédiatrique
La sécurité et l'efficacité du paclitaxel sous forme de nanoparticules liées à l'albumine chez les enfants et adolescents âgés de 0 à moins de 18 ans n'ont pas été établies. Les données actuellement disponibles sont décrites aux rubriques Effets indésirables, Propriétés pharmacodynamiques et Propriétés pharmacocinétiques mais aucune recommandation sur la posologie ne peut être donnée. Il n'existe pas d'utilisation justifiée de BUGVI dans la population pédiatrique pour l'indication du cancer du sein métastatique, de l'adénocarcinome du pancréas métastatique ou du cancer bronchique non à petites cellules.
Mode d'administration
Administrer la dispersion reconstituée de BUGVI par voie intraveineuse à l'aide d'un kit de perfusion muni d'un filtre de 15 μm. Après l'administration, il est recommandé de rincer la tubulure avec une solution injectable de chlorure de sodium à 0,9 mg/mL (9 %) afin de garantir l'administration de la dose complète.
Pour les instructions concernant la reconstitution du médicament avant administration, voir la rubrique Précautions particulières d’élimination et de manipulation.
Durée de conservation :
Flacons non ouverts
3 ans
Stabilité de la dispersion reconstituée dans le flacon
La stabilité physico-chimique en cours d'utilisation a été démontrée pendant 24 heures à une température comprise entre 2 °C et 8 °C dans l'emballage d'origine, à l'abri de la lumière.
Stabilité de la dispersion reconstituée dans la poche pour perfusion
La stabilité physico-chimique en cours d'utilisation a été démontrée pour une période de 24 heures à une température comprise entre 2 °C et 8 °C, suivie d'une période de 4 heures à 25 °C, à l'abri de la lumière.
Cependant, du point de vue microbiologique, à moins que la méthode de reconstitution et de remplissage des poches pour perfusion n'exclue les risques de contamination microbienne, le produit doit être utilisé immédiatement après la reconstitution et le remplissage des poches pour perfusion.
S'il n'est pas utilisé immédiatement, la durée et les conditions de conservation en cours d'utilisation relèvent de la responsabilité de l'utilisateur.
La durée totale de conservation combinée du médicament reconstitué dans le flacon et dans la poche pour perfusion, lorsqu'il est réfrigéré et à l'abri de la lumière, est de 24 heures. Ensuite, le médicament peut être conservé dans la poche pour perfusion pendant 4 heures, à une température ne dépassant pas 25 °C.
Précautions particulières de conservation :
Flacons non ouverts
Conserver le flacon dans l'emballage extérieur à l'abri de la lumière. Ni la congélation ni la réfrigération n'affectent la stabilité du produit. Ce médicament ne nécessite pas de précautions particulières de conservation concernant la température.
Dispersion reconstituée
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique Durée de conservation.
Ce médicament ne doit pas être mélangé avec d'autres médicaments à l'exception de ceux mentionnés dans la rubrique Précautions particulières d’élimination et de manipulation.
Il n'existe pas d'antidote connu en cas de surdosage de paclitaxel. En cas de surdosage, le patient doit faire l'objet d'une surveillance étroite. Le traitement doit viser les principales complications prévisibles d'un surdosage que sont l'aplasie médullaire, la mucite et la neuropathie périphérique.
Classe pharmacothérapeutique : Antinéoplasiques, alcaloïdes végétaux et autres médicaments d'origine naturelle, taxanes, code ATC : L01CD01.
Mécanisme d'action
Le paclitaxel est un agent antimicrotubule qui stimule l'assemblage des dimères de tubuline en microtubules et stabilise ces derniers en empêchant leur dépolymérisation. Cette stabilité inhibe la réorganisation dynamique normale du réseau de microtubules, un phénomène essentiel aux fonctions vitales des cellules au cours de l'interphase et de la mitose. De plus, le paclitaxel induit la formation anormale de groupements ou de faisceaux de microtubules pendant toute la durée du cycle cellulaire ainsi que la constitution de multiples asters de microtubules pendant la mitose.
BUGVI contient des nanoparticules de paclitaxel-albumine sérique humaine d'une taille d'environ 130 nm, le paclitaxel étant à l'état amorphe, non cristallin. Après administration intraveineuse, les nanoparticules se dissocient rapidement en complexes solubles de paclitaxel lié à l'albumine d'une taille d'environ 10 nm. L'albumine est connue pour faciliter la transcytose endothéliale par les cavéoles de composants plasmatiques et des études in vitro ont démontré que sa présence dans le paclitaxel sous forme de nanoparticules liées à l'albumine favorise le transport du paclitaxel à travers les cellules endothéliales. L'hypothèse est avancée selon laquelle ce transport transendothélial cavéolaire facilité fait intervenir le récepteur de l'albumine gp60, et l'accumulation de paclitaxel est augmentée dans la zone tumorale grâce à la protéine SPARC (Secreted Protein Acidic and Rich in Cysteine) qui se lie à l'albumine.
Efficacité et sécurité clinique
Cancer du sein
Des données provenant de 106 patients ayant participé à deux études en ouvert à bras unique, et de 454 patients traités au cours d'une étude comparative randomisée de phase III, sont disponibles pour soutenir l'utilisation du paclitaxel sous forme de nanoparticules liées à l'albumine dans le traitement du cancer du sein métastatique. Ces informations sont présentées ci-dessous.
Etudes en ouvert à bras unique
Dans une étude, le paclitaxel sous forme de nanoparticules liées à l'albumine a été administré par perfusion de 30 minutes à la dose de 175 mg/m2 à 43 patients atteints d'un cancer du sein métastatique. Lors du second essai, une dose de 300 mg/m2 a été administrée par perfusion de 30 minutes chez 63 patients atteints d'un cancer du sein métastatique. Les patients n'avaient pas reçu de prémédication par corticoïdes ni de traitement par G-CSF. Les cycles étaient administrés à 3 semaines d'intervalle. Les taux de réponse observés chez tous les patients étaient respectivement de 39,5 % (IC à 95 % : 24,9 %-54,2 %) et de 47,6 % (IC à 95 % : 35,3 %-60,0 %). Le délai médian jusqu'à progression de la maladie était de 5,3 mois (175 mg/m2 ; IC à 95 % : 4,6-6,2 mois) et de 6,1 mois (300 mg/m2 ; IC à 95 % : 4,2-9,8 mois).
Etude comparative randomisée
Cet essai multicentrique a été mené chez des patients atteints d'un cancer du sein métastatique traités toutes les 3 semaines avec du paclitaxel en monothérapie, soit par paclitaxel à base de solvant à la dose de 175 mg/m2 administré en perfusion de 3 heures avec une prémédication pour prévenir le risque d'hypersensibilité (N = 225), soit par paclitaxel sous forme de nanoparticules liées à l'albumine à la dose de 260 mg/m2 administré en perfusion de 30 minutes sans prémédication (N = 229).
Soixante-quatre pour cent des patients présentaient un indice de performance altéré (ECOG 1 ou 2) à l'entrée dans l'étude ; 79 % d'entre eux présentaient des métastases viscérales ; et 76 % présentaient > 3 sites métastatiques. Quatorze pour cent des patients n'avaient jamais été soumis à une chimiothérapie antérieure ; 27 % uniquement avaient reçu une chimiothérapie en traitement adjuvant, 40 % uniquement une chimiothérapie en traitement métastatique et 19 % dans les deux situations (chimiothérapie adjuvante et métastatique). Cinquante-neuf pour cent des patients avaient reçu le médicament à l'étude au moins en deuxième ligne de traitement. Soixante-dix-sept pour cent des patients avaient été antérieurement exposés aux anthracyclines.
Les résultats du taux de réponse globale et du délai jusqu'à la progression de la maladie, et ceux de la survie sans progression de la maladie et de la survie pour les patients ayant reçu > 1 première ligne de traitement, sont présentés ci-dessous.
Tableau 8 : Taux de réponse globale, délai médian jusqu'à la progression de la maladie et survie sans progression de la maladie estimés par l'investigateur
|
Variable d'efficacité |
Paclitaxel sous forme de nanoparticules liées à l'albumine (260 mg/m2) |
Paclitaxel à base de solvant (175 mg/m2) |
Valeur de p |
|
Taux de réponse [IC à 95 %] (%) |
|||
|
> 1re ligne de traitement |
26,5 [18,98 ; 34,05] (n = 132) |
13,2 [7,54 ; 18,93] (n = 136) |
0,006a |
|
* Délai médian jusqu'à progression de la maladie [IC à 95 %] (semaines) |
|||
|
> 1re ligne de traitement |
20,9 [15,7 ; 25,9] (n = 131) |
16,1 [15,0 ; 19,3] (n = 135) |
0,011b |
|
* Survie médiane sans progression [IC à 95 %] (semaines) |
|||
|
> 1re ligne de traitement |
20,6 [15,6 ; 25,9] (n = 131) |
16,1 [15,0 ; 18,3] (n = 135) |
0,010b |
|
* Survie [IC à 95 %] (semaines) |
|||
|
> 1re ligne de traitement |
56,4 [45,1 ; 76,9] (n = 131) |
46,7 [39,0 ; 55,3] (n = 136) |
0,020b |
* Ces données sont basées sur le rapport d'étude clinique : CA012-0 Addendum final (23 mars 2005)a Test du Chi2b Test du log-rank
La sécurité du paclitaxel sous forme de nanoparticules liées à l'albumine a été évaluée chez deux cent vingt-neuf patients ayant reçu le produit au cours de cet essai clinique randomisé et contrôlé. La neurotoxicité liée au paclitaxel a été évaluée par l'amélioration d'un grade chez les patients souffrant d'une neuropathie périphérique de grade 3, et ce à tout moment au cours du traitement. L'évolution de la neuropathie périphérique due à une toxicité cumulative du paclitaxel sous forme de nanoparticules liées à l'albumine après > 6 cycles de traitement vers un retour à l'état initial n'a pas été évaluée et reste inconnue.
Adénocarcinome du pancréas
Une étude multicentrique, internationale, randomisée, en ouvert a été menée chez 861 patients pour comparer l'association paclitaxel sous forme de nanoparticules liées à l'albumine/gemcitabine versus gemcitabine en monothérapie en traitement de première ligne de l'adénocarcinome du pancréas métastatique. Les patients (N = 431) ont reçu le paclitaxel sous forme de nanoparticules liées à l'albumine en perfusion intraveineuse de 30 à 40 minutes à la dose de 125 mg/m2, suivi de la gemcitabine en perfusion intraveineuse de 30 à 40 minutes à la dose de 1 000 mg/m2, administrée les Jours 1, 8 et 15 de chaque cycle de 28 jours. Dans le bras de traitement comparateur, les patients (N = 430) ont reçu la gemcitabine en monothérapie conformément à la dose et au schéma posologique recommandés. Le traitement était administré jusqu'à la progression de la maladie ou jusqu'au développement d'une toxicité inacceptable. Chez les 431 patients présentant un adénocarcinome du pancréas ayant été randomisés pour recevoir le paclitaxel sous forme de nanoparticules liées à l'albumine en association avec la gemcitabine, les patients étaient en majorité blancs (93 %), 4 % étaient noirs et 2 % étaient asiatiques. L'indice de Karnofsky (KPS) était de 100 chez 16 % des patients, de 90 chez 42 %, de 80 chez 35 %, de 70 chez 7 % et inférieur à 70 chez < 1 % des patients. Les patients présentant un risque cardiovasculaire élevé, des antécédents d'artériopathie périphérique et/ou de collagénose et/ou de pneumopathie interstitielle n'ont pas été inclus dans l'étude.
La durée de traitement médiane a été de 3,9 mois dans le bras paclitaxel sous forme de nanoparticules liées à l'albumine/gemcitabine et de 2,8 mois dans le bras gemcitabine. 32 % des patients du bras paclitaxel sous forme de nanoparticules liées à l'albumine/gemcitabine versus 15 % des patients du bras gemcitabine ont reçu 6 mois de traitement ou plus. Dans la population traitée, la dose-intensité relative médiane de la gemcitabine a été de 75 % dans le bras paclitaxel sous forme de nanoparticules liées à l'albumine/gemcitabine et de 85 % dans le bras gemcitabine. La dose-intensité relative médiane de paclitaxel sous forme de nanoparticules liées à l'albumine a été de 81 %. La dose cumulée médiane administrée a été plus élevée dans le bras paclitaxel sous forme de nanoparticules liées à l'albumine/gemcitabine (11 400 mg/m2) que dans le bras gemcitabine (9 000 mg/m2).
Le critère principal d'évaluation de l'efficacité a été la survie globale (SG). Les principaux critères secondaires ont été la survie sans progression (SSP) et le taux de réponse globale (TRG), tous deux évalués par une analyse radiologique indépendante centralisée en aveugle selon les critères RECIST (version 1.0).
Tableau 9 : Résultats d'efficacité de l'étude randomisée chez des patients présentant un adénocarcinome du pancréas (population en intention de traiter)
|
|
Paclitaxel sous forme de nanoparticules liées à l'albumine (125 mg/m2)/gemcitabine (N = 431) |
Gemcitabine (N = 430) |
|
Survie globale |
||
|
Nombre de décès (%) |
333 (77) |
359 (83) |
|
Survie globale médiane, mois (IC à 95 %) |
8,5 (7,89 ; 9,53) |
6,7 (6,01 ; 7,23) |
|
RRA+G/G (IC à 95 %)a |
0,72 (0,617 ; 0,835) |
|
|
Valeur de pb |
< 0,0001 |
|
|
Taux de survie en % (IC à 95 %) à |
|
|
|
1 an |
35 % (29,7 ; 39,5) |
22 % (18,1 ; 26,7) |
|
2 ans |
9 % (6,2 ; 13,1) |
4 % (2,3 ; 7,2) |
|
75e percentile de survie globale (mois) |
14,8 |
11,4 |
|
Survie sans progression |
||
|
Décès ou progression, n (%) |
277 (64) |
265 (62) |
|
Survie sans progression médiane, mois (IC à 95 %) |
5,5 (4,47 ; 5,95) |
3,7 (3,61 ; 4,04) |
|
HRA+G/G (IC à 95 %)a |
0,69 (0,581 ; 0,821) |
|
|
Valeur de pb |
< 0,0001 |
|
|
Taux de réponse globale |
||
|
Réponse globale complète ou partielle confirmée, n (%) |
99 (23) |
31 (7) |
|
IC à 95 % |
19,1 ; 27,2 |
5,0 ; 10,1 |
|
pA+G/pG (IC à 95 %) |
3,19 (2,178 ; 4,662) |
|
|
Valeur de p (test du Chi2) |
< 0,0001 |
|
IC = intervalle de confiance, RRA+G/G = rapport de risque du paclitaxel sous forme de nanoparticules liées à l'albumine+gemcitabine/gemcitabine, pA+G/pG = rapport des taux de réponse du paclitaxel sous forme de nanoparticules liées à l'albumine+gemcitabine/gemcitabine.
a Modèle de risques proportionnels de Cox stratifié.
b Test du log-rank stratifié par région géographique (Amérique du Nord versus autres régions), KPS (70 à 80 versus 90 à 100) et présence de métastases hépatiques (oui versus non).
Une amélioration statistiquement significative de la SG a été observée chez les patients traités par paclitaxel sous forme de nanoparticules liées à l'albumine/gemcitabine par rapport à la gemcitabine en monothérapie, avec une augmentation de 1,8 mois de la SG médiane, une réduction globale de 28 % du risque de décès, une augmentation de 59 % du taux de survie à 1 an et une augmentation de 125 % du taux de survie à 2 ans.
Figure 1 : Courbe de Kaplan-Meier de la survie globale (population en intention de traiter)
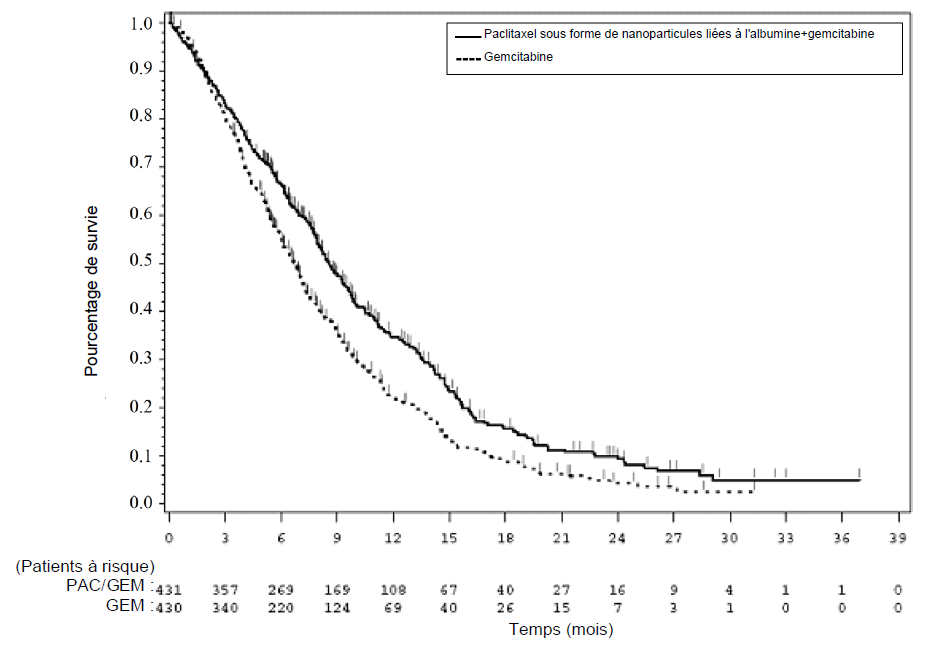
Les effets du traitement sur la SG ont été en faveur du bras paclitaxel sous forme de nanoparticules liées à l'albumine/gemcitabine dans la majorité des sous-groupes prédéfinis (y compris sexe, KPS, région géographique, siège primaire du cancer du pancréas, stade au moment du diagnostic, présence de métastases hépatiques, présence d'une carcinose péritonéale, antécédents de duodénopancréatectomie céphalique (intervention de Whipple), présence d'un stent biliaire à l'inclusion, présence de métastases pulmonaires et nombre de sites métastatiques). Chez les patients âgés de ≥ 75 ans des bras paclitaxel sous forme de nanoparticules liées à l'albumine/gemcitabine et gemcitabine, le rapport de risque (RR) de survie a été de 1,08 (IC à 95 % : 0,653 ; 1,797). Chez les patients ayant un taux de CA 19-9 normal à l'inclusion, le RR de survie a été de 1,07 (IC à 95 % : 0,692 ; 1,661).
Une amélioration statistiquement significative de la SSP a été observée chez les patients traités par paclitaxel sous forme de nanoparticules liées à l'albumine/gemcitabine par rapport à la gemcitabine en monothérapie, avec une augmentation de 1,8 mois de la SSP médiane.
Cancer bronchique non à petites cellules
Une étude multicentrique, randomisée, en ouvert a été menée chez 1 052 patients présentant un cancer bronchique non à petites cellules de stade IIIb/IV naïfs de chimiothérapie. L'étude visait à comparer le paclitaxel sous forme de nanoparticules liées à l'albumine en association avec le carboplatine par rapport aupaclitaxel à base de solvant en association avec le carboplatine en traitement de première ligne chez des patients présentant un cancer bronchique non à petites cellules avancé. Plus de 99 % des patients avaient un indice de performance ECOG (Eastern Cooperative Oncology Group) de 0 ou 1.
Les patients présentant une neuropathie préexistante de grade ≥ 2 ou des facteurs de risque médicaux graves impliquant l'un des systèmes d'organes majeurs n'ont pas été inclus. Les patients (N = 521) ont reçu le paclitaxel sous forme de nanoparticules liées à l'albumine en perfusion intraveineuse de 30 minutes à la dose de 100 mg/m2 les Jours 1, 8 et 15 de chaque cycle de 21 jours, sans prémédication par corticoïdes et sans traitement prophylactique par G-CSF. Immédiatement après la fin de la perfusion de paclitaxel sous forme de nanoparticules liées à l'albumine, le carboplatine a été administré par voie intraveineuse à la dose ASC = 6 mg•min/mL le Jour 1 uniquement de chaque cycle de 21 jours. Les patients (N = 531) ont reçu le paclitaxel à base de solvant en perfusion intraveineuse de 3 heures à la dose de 200 mg/m2 avec la prémédication habituelle, suivi immédiatement du carboplatine administré par voie intraveineuse à la dose ASC = 6 mg•min/mL. Chaque médicament était administré le Jour 1 de chaque cycle de 21 jours. Dans les deux bras de l'étude, le traitement a été administré jusqu'à la progression de la maladie ou survenue d'une toxicité inacceptable. Dans les deux bras de l'étude, les patients ont reçu un nombre médian de 6 cycles de traitement.
Le critère principal d'évaluation de l'efficacité était le taux de réponse globale, défini comme le pourcentage de patients ayant obtenu une réponse objective, complète ou partielle, confirmée par l'analyse radiologique en aveugle, indépendante et centralisée selon les critères RECIST (version 1.0). Les patients du bras paclitaxel sous forme de nanoparticules liées à l'albumine/carboplatine ont présenté un taux de réponse globale significativement plus élevé que les patients du bras témoin : 33 % versus 25 %, p = 0,005 (tableau 10). Une différence significative du taux de réponse globale a été observée dans le bras paclitaxel sous forme de nanoparticules liées à l'albumine/carboplatine par rapport au bras témoin chez les patients présentant un cancer bronchique non à petites cellules d'histologie épidermoïde (N = 450, 41 % versus 24 %, p < 0,001). Cependant, cette différence ne s'est pas traduite en différence de SSP ou de SG. Aucune différence n'a été observée en termes de TRG entre les bras de traitement chez les patients présentant un cancer bronchique non à petites cellules d'histologie non épidermoïde (N = 602, 26 % versus 25 %, p = 0,808).
Tableau 10 : Taux de réponse globale dans l'étude randomisée menée dans le cancer bronchique non à petites cellules (population en intention de traiter)
|
Paramètre d'efficacité |
Paclitaxel sous forme de nanoparticules liées à l'albumine (100 mg/m2/semaine)+carboplatine (N = 521) |
Paclitaxel à base de solvant (200 mg/m2 toutes les 3 semaines)+carboplatine (N = 531) |
|
Taux de réponse globale (analyse indépendante) |
||
|
Réponse globale complète ou partielle confirmée, n (%) |
170 (33 %) |
132 (25 %) |
|
IC à 95 % (%) |
28,6 ; 36,7 |
21,2 ; 28,5 |
|
pA/pT (IC à 95,1 %) |
1,313 (1,082 ; 1,593) |
|
|
Valeur de pa |
0,005 |
|
IC = intervalle de confiance ; RRA/T = rapport de risque du paclitaxel sous forme de nanoparticules liées à l'albumine+carboplatine/paclitaxel à base de solvant+carboplatine ; pA/pT = rapport des taux de réponse du paclitaxel sous forme de nanoparticules liées à l'albumine+carboplatine/paclitaxel à base de solvant+carboplatine.
a Valeur de p basée sur un test du Chi2.
Aucune différence statistiquement significative n'a été observée entre les deux bras de traitement en termes de survie sans progression (selon l'évaluation radiologique en aveugle) et de survie globale. Une analyse de non-infériorité a été effectuée pour la SSP et la SG avec une marge de non-infériorité prédéfinie de 15 %. Le critère de non-infériorité a été atteint pour la SSP et la SG, la limite supérieure de l'intervalle de confiance à 95 % des rapports de risque associés étant inférieure à 1,176 (tableau 11).
Tableau 11 : Analyses de non-infériorité de la survie sans progression et de la survie globale dans l'étude randomisée menée sur le cancer bronchique non à petites cellules (population en intention de traiter)
|
Paramètre d'efficacité |
Paclitaxel sous forme de nanoparticules liées à l'albumine (100 mg/m2/semaine)+carboplatine (N = 521) |
Paclitaxel à base de solvant (200 mg/m2 toutes les 3 semaines)+carboplatine (N = 531) |
|
Survie sans progressiona (analyse indépendante) |
||
|
Décès ou progression, n (%) |
429 (82 %) |
442 (83 %) |
|
SSP médiane (IC à 95 %) (mois) |
6,8 (5,7 ; 7,7) |
6,5 (5,7 ; 6,9) |
|
RRA/T (IC à 95 %) |
0,949 (0,830 ; 1,086) |
|
|
Survie globale |
||
|
Nombre de décès, n (%) |
360 (69 %) |
384 (72 %) |
|
SG médiane (IC à 95 %) (mois) |
12,1 (10,8 ; 12,9) |
11,2 (10,3 ; 12,6) |
|
RRA/T (IC à 95,1 %) |
0,922 (0,797 ; 1,066) |
|
IC = intervalle de confiance ; RRA/T = rapport de risque du paclitaxel sous forme de nanoparticules liées à l'albumine+carboplatine/paclitaxel à base de solvant+carboplatine ; pA/pT = rapport des taux de réponse du paclitaxel sous forme de nanoparticules liées à l'albumine+carboplatine/paclitaxel à base de solvant+carboplatine.
a Conformément à la ligne directrice de l'EMA relative aux aspects méthodologiques pour l'utilisation du critère de SSP, les observations manquantes ou l'instauration d'un nouveau traitement ultérieur n'ont pas été censurées.
Population pédiatrique
La sécurité et l'efficacité chez les patients pédiatriques n'ont pas été établies (voir rubrique Posologie et mode d'administration).
L'étude ABI-007-PST-001, une étude de détermination de dose de phase I/II, multicentrique et en ouvert, visant à évaluer la sécurité, la tolérance et l'efficacité préliminaire du paclitaxel sous forme de nanoparticules liées à l'albumine administré une fois par semaine chez des patients pédiatriques atteints de tumeurs solides en rechute ou réfractaires, comprenait un total de 106 patients âgés de ≥ 6 mois à ≤ 24 ans.
La phase I de l'étude comprenait un total de 64 patients âgés de 6 mois à moins de 18 ans et a permis de déterminer la dose maximale tolérée (DMT) de 240 mg/m2, administrée par perfusion intraveineuse de 30 minutes, les Jours 1, 8 et 15 de chaque cycle de 28 jours.
Dans la phase II, un total de 42 patients âgés de 6 mois à 24 ans et atteints d'un sarcome d'Ewing, d'un neuroblastome ou d'un rhabdomyosarcome en rechute ou réfractaire ont été inclus selon un plan minimax en deux étapes de Simon afin d'évaluer l'activité antitumorale en fonction du taux de réponse globale (TRG). Sur ces 42 patients, un patient était âgé de < 2 ans, 27 patients étaient âgés de ≥ 2 à < 12 ans, 12 patients étaient âgés de ≥ 12 à < 18 ans et 2 patients adultes étaient âgés de ≥ 18 à 24 ans.
Les patients ont reçu un traitement à la DMT d'une durée médiane de 2 cycles. Sur les 41 patients éligibles pour une évaluation de l'efficacité à la phase I, 1 patient du groupe rhabdomyosarcome (N = 14) a présenté une réponse partielle (RP) confirmée, se traduisant par un TRG de 7,1 % (IC à 95 % : 0,2 ; 33,9). Aucune réponse complète (RC) confirmée ni aucune RP n'ont été observées, que ce soit dans le groupe sarcome d'Ewing (N = 13) ou dans le groupe neuroblastome (N = 14). Aucun des bras de l'étude n'est passé à la phase II car le protocole définissait une exigence de ≥ 2 patients présentant une réponse confirmée, et cette exigence n'était pas satisfaite.
La survie globale médiane, qui tient compte de la période de suivi de 1 an, était de 32,1 semaines pour le groupe sarcome d'Ewing (IC à 95 % : 21,4 ; 72,9), de 32,0 semaines pour le groupe neuroblastome (IC à 95 % : 12 ; non établi) et de 19,6 semaines pour le groupe rhabdomyosarcome (IC à 95 % : 4 ; 25,7).
Le profil de sécurité global du paclitaxel sous forme de nanoparticules liées à l'albumine chez les patients pédiatriques était conforme avec le profil de sécurité connu du paclitaxel sous forme de nanoparticules liées à l'albumine chez l'adulte (voir rubrique Effets indésirables). Ces résultats ont mené à la conclusion selon laquelle le paclitaxel sous forme de nanoparticules liées à l'albumine en monothérapie n'apporte pas un bénéfice significatif en termes d'activité clinique ou de survie qui justifierait la poursuite de son développement dans la population pédiatrique.
La pharmacocinétique du paclitaxel total suite à des perfusions de 30 et de 180 minutes de paclitaxel sous forme de nanoparticules liées à l'albumine à des paliers de dose allant de 80 à 375 mg/m2 a été déterminée au cours d'études cliniques. L'exposition au paclitaxel (ASC) augmentait linéairement de 2 653 à 16 736 ng.h/mL après l'administration de doses allant de 80 à 300 mg/m2.
Lors d'une étude menée chez des patients atteints de tumeurs solides à un stade avancé, les caractéristiques pharmacocinétiques du paclitaxel suite à l'administration du paclitaxel sous forme de nanoparticules liées à l'albumine par voie intraveineuse à la dose de 260 mg/m2 en perfusion de 30 minutes ont été comparées avec celles obtenues suite à la perfusion de 175 mg/m2 de paclitaxel à base de solvant administrée en 3 heures. Selon une analyse pharmacocinétique non compartimentale, la clairance plasmatique du paclitaxel mesurée sous paclitaxel sous forme de nanoparticules liées à l'albumine était plus importante (43 %) que celle suivant l'injection de paclitaxel à base de solvant et son volume de distribution était également plus élevé (53 %). Aucune différence n'a été observée au niveau des demi-vies d'élimination terminale.
Lors d'une étude à doses répétées chez 12 patients recevant le paclitaxel sous forme de nanoparticules liées à l'albumine par voie intraveineuse à la dose de 260 mg/m2, la variabilité intra-patient de l'ASC était de 19 % (intervalle = 3,21 %-37,70 %). Aucun signe d'accumulation du paclitaxel n'a été observé lors des multiples cures de traitement.
Distribution
Après administration du paclitaxel sous forme de nanoparticules liées à l'albumine chez des patients présentant des tumeurs solides, le paclitaxel est distribué uniformément dans les érythrocytes et dans le plasma et est fortement lié aux protéines plasmatiques (94 %).
La liaison aux protéines du paclitaxel après l'administration de paclitaxel sous forme de nanoparticules liées à l'albumine a été évaluée par ultrafiltration dans une étude de comparaison intra-patient. La fraction de paclitaxel libre était significativement plus élevée avec le paclitaxel sous forme de nanoparticules liées à l'albumine (6,2 %) qu'avec la formulation paclitaxel à base de solvant (2,3 %). Par conséquent, l'exposition au paclitaxel non lié a été plus importante avec le paclitaxel sous forme de nanoparticules liées à l'albumine qu'avec le paclitaxel à base de solvant, malgré une exposition totale comparable. Cela pourrait s'expliquer par le fait que le paclitaxel n'est pas piégé dans les micelles de Cremophor EL comme avec le paclitaxel à base de solvant. D'après la littérature publiée, les études effectuées in vitro sur des protéines sériques humaines (utilisant des concentrations de paclitaxel allant de 0,1 à 50 μg/mL) indiquent que le taux de liaison protéique du paclitaxel n'est pas modifié en présence de cimétidine, de ranitidine, de dexaméthasone ou de diphénhydramine.
Selon l'analyse pharmacocinétique de population, le volume de distribution total est d'environ 1 741 litres ; le grand volume de distribution indique une distribution extravasculaire et/ou une fixation tissulaire importantes du paclitaxel.
Biotransformation et élimination
D'après la littérature publiée, les études menées in vitro sur des microsomes de foie humain et différentes coupes de tissu indiquent que le paclitaxel est principalement métabolisé en 6α-hydroxypaclitaxel et en deux métabolites mineurs, 3'-p-hydroxypaclitaxel et 6α-3'-p-dihydroxypaclitaxel. La formation de ces métabolites hydroxylés est catalysée par les isoenzymes CYP2C8, CYP3A4 et de façon concomitante par CYP2C8 et CYP3A4, respectivement.
Chez des patients atteints d'un cancer du sein métastatique, après une perfusion de 30 minutes de paclitaxel sous forme de nanoparticules liées à l'albumine à la dose de 260 mg/m2, la valeur moyenne de l'excrétion urinaire cumulée de substance active sous forme inchangée s'élevait à 4 % de la dose totale administrée, avec moins de 1 % sous forme de métabolites 6α-hydroxypaclitaxel et 3'-p-hydroxypaclitaxel, ce qui indique l'importance de la clairance non rénale. Le paclitaxel est éliminé principalement par métabolisme hépatique et excrétion biliaire.
Dans l'éventail de doses cliniques allant de 80 à 300 mg/m2, la clairance plasmatique moyenne du paclitaxel est de 13 à 30 L/h/m2 et la demi-vie terminale moyenne de 13 à 27 heures.
Insuffisance hépatique
L'effet de l'insuffisance hépatique sur la pharmacocinétique de population du paclitaxel sous forme de nanoparticules liées à l'albumine a été étudié chez des patients atteints de tumeurs solides à un stade avancé. Cette analyse incluait des patients présentant une fonction hépatique normale (n = 130) et des patients présentant une insuffisance hépatique préexistante légère (n = 8), modérée (n = 7) ou sévère (n = 5) (selon les critères de l'Organ Dysfunction Working Group du NCI). Les résultats révèlent que l'insuffisance hépatique légère (bilirubine totale > 1 et ≤ 1,5 × LSN) n'a pas d'effet cliniquement important sur la pharmacocinétique du paclitaxel. Chez les patients présentant une insuffisance hépatique modérée (bilirubine totale > 1,5 et ≤ 3 × LSN) ou sévère (bilirubine totale > 3 et ≤ 5 × LSN), la vitesse d'élimination maximale du paclitaxel est diminuée de 22 % à 26 % et l'ASC moyenne augmentée d'environ 20 % par rapport aux patients présentant une fonction hépatique normale. L'insuffisance hépatique n'a pas d'effet sur la Cmax moyenne du paclitaxel. De plus, l'élimination du paclitaxel présente une corrélation inverse avec la bilirubine totale et une corrélation positive avec l'albuminémie.
La modélisation pharmacocinétique/pharmacodynamique indique qu'il n'y a pas de corrélation entre la fonction hépatique (indiquée par le taux initial d'albumine ou de bilirubine totale) et la neutropénie après ajustement pour l'exposition au paclitaxel sous forme de nanoparticules liées à l'albumine.
Aucune donnée pharmacocinétique n'est disponible chez les patients présentant un taux de bilirubine totale > 5 × LSN ou chez les patients atteints d'un adénocarcinome du pancréas métastatique (voir rubrique Posologie et mode d'administration).
Insuffisance rénale
L'analyse pharmacocinétique de population incluait des patients présentant une fonction rénale normale (n = 65) et des patients présentant une insuffisance rénale préexistante légère (n = 61), modérée (n = 23) ou sévère (n = 1) (conformément aux critères 2010 du projet de ligne directrice [Draft guidance] de la FDA). L'insuffisance rénale légère à modérée (clairance de la créatinine ≥ 30 et < 90 mL/min) n'a pas d'effet cliniquement important sur la vitesse d'élimination maximale et l'exposition systémique (ASC et Cmax) du paclitaxel. Les données pharmacocinétiques ne sont pas suffisantes chez les patients présentant une insuffisance rénale sévère et aucune donnée n'est disponible chez les patients présentant une insuffisance rénale terminale.
Personnes âgées
L'analyse pharmacocinétique de population du paclitaxel sous forme de nanoparticules liées à l'albumine incluait des patients âgés de 24 à 85 ans et révèle que l'âge n'a pas d'influence significative sur la vitesse d'élimination maximale et l'exposition systémique (ASC et Cmax) du paclitaxel.
Une modélisation pharmacocinétique/pharmacodynamique utilisant les données de 125 patients présentant des tumeurs solides avancées indique que les patients âgés de ≥ 65 ans peuvent être plus susceptibles de développer une neutropénie pendant le premier cycle de traitement, bien que l'âge n'ait pas d'effet sur l'exposition plasmatique au paclitaxel.
Population pédiatrique
La pharmacocinétique du paclitaxel après une administration intraveineuse de 30 minutes à des paliers de dose de 120 mg/m2 à 270 mg/m2 a été déterminée chez 64 patients (2 à ≤ 18 ans) au cours de la phase I d'une étude de phase I/II menée chez des patients pédiatriques atteints de tumeurs solides en rechute ou réfractaires. Après une augmentation de la dose de 120 à 270 mg/m2, l'ASC(0-inf) et la Cmax moyennes du paclitaxel variaient de 8 867 à 14 361 ng.h/mL et de 3 488 à 8 078 ng/mL, respectivement.
Les valeurs d'exposition au médicament maximales normalisées en fonction de la dose étaient comparables dans la gamme de doses étudiées. Cependant, les valeurs d'exposition au médicament totales normalisées en fonction de la dose étaient comparables uniquement entre 120 mg/m2 et 240 mg/m2, avec l'ASC∞ normalisée en fonction de la dose la plus basse pour le palier de dose de 270 mg/m2. A la DMT de 240 mg/m2, la clairance moyenne était de 19,1 L/h et la demi-vie terminale moyenne était de 13,5 heures.
Chez les enfants et les adolescents, l'exposition au paclitaxel augmentait avec l'administration de doses plus élevées et les expositions hebdomadaires au médicament étaient plus élevées que chez les patients adultes.
Autres facteurs intrinsèques
Les analyses pharmacocinétiques de population du paclitaxel sous forme de nanoparticules liées à l'albumine indiquent que le sexe, l'origine ethnique (Asiatiques versus Blancs) et le type de tumeur solide n'ont pas d'effet cliniquement important sur l'exposition systémique (ASC et Cmax) du paclitaxel. L'ASC du paclitaxel est diminuée d'environ 25 % chez les patients pesant 50 kg par rapport aux patients pesant 75 kg. La pertinence clinique de cette observation n'est pas certaine.
Le paclitaxel a une influence mineure à modérée sur l'aptitude à conduire des véhicules et à utiliser des machines. Le paclitaxel peut provoquer des effets indésirables tels que de la fatigue (très fréquent) et des sensations vertigineuses (fréquent) qui peuvent affecter l'aptitude à conduire des véhicules et à utiliser des machines. Il convient de conseiller aux patients de ne pas conduire de véhicules et de ne pas utiliser des machines s'ils présentent une fatigue ou des sensations vertigineuses.
Le potentiel cancérogène du paclitaxel n'a pas été étudié. Cependant, selon les données publiées dans la littérature, le paclitaxel est un agent potentiellement cancérogène et génotoxique aux doses utilisées en clinique en raison de son mécanisme d'action pharmacodynamique. Le paclitaxel s'est révélé clastogène in vitro (aberrations chromosomiques dans les lymphocytes humains) et in vivo (test du micronucleus chez la souris). Le paclitaxel s'est révélé génotoxique in vivo (test du micronucleus chez la souris) mais ne s'est pas révélé mutagène dans le test d'Ames ni dans celui de mutation génique dans des cellules d'ovaire de hamster chinois/hypoxanthine guanine phosphoribosyltransférase (CHO/HGPRT).
Le paclitaxel administré à des doses inférieures à la dose thérapeutique humaine est associé à une baisse de la fertilité lorsqu'il est administré avant et pendant la période d'accouplement chez des rats mâles et femelles et à une foetotoxicité chez le rat. Les études effectuées chez l'animal ont révélé des effets toxiques, non réversibles sur les organes reproductifs mâles à des niveaux d'exposition similaires à ceux observés en clinique.
Le paclitaxel et/ou ses métabolites sont excrétés dans le lait de rates allaitantes. Après administration intraveineuse de paclitaxel radiomarqué à des rates aux Jours 9 et 10 du postpartum, les concentrations de radioactivité dans le lait étaient plus élevées que dans le plasma et ont diminué parallèlement aux concentrations plasmatiques.
Précautions à prendre pour la préparation et l'administration
Le paclitaxel est un médicament anticancéreux cytotoxique et, comme tout composé potentiellement toxique, le paclitaxel doit être manipulé avec prudence. Le port de gants, lunettes et vêtements de protection est recommandé. En cas de contact de la dispersion avec la peau, laver la peau immédiatement et abondamment avec de l'eau et du savon. En cas de contact avec les muqueuses, rincer abondamment avec de l'eau. Le paclitaxel ne doit être préparé et administré que par un personnel dûment formé à la manipulation d'agents cytotoxiques. Les femmes enceintes de l'équipe soignante ne doivent pas manipuler le paclitaxel.
En raison du risque d'extravasation, il est conseillé de surveiller attentivement le site de perfusion afin de déceler toute infiltration éventuelle pendant l'administration du médicament. Le fait de limiter la durée de la perfusion du paclitaxel à 30 minutes, conformément aux instructions, permet de réduire le risque de réactions liées à la perfusion.
Reconstitution et administration du produit
BUGVI est commercialisé sous forme de poudre lyophilisée stérile à reconstituer avant utilisation. Après reconstitution, chaque mL de dispersion contient 5 mg de paclitaxel dans une formulation de nanoparticules liées à l'albumine.
Flacon de 100 mg : à l'aide d'une seringue stérile, 20 mL de solution pour perfusion de chlorure de sodium à 0,9 mg/mL (9 %) doivent être injectés lentement (pendant au moins 1 minute) dans un flacon de BUGVI.
La solution doit être dirigée vers la paroi interne du flacon. Afin d'éviter tout risque de formation de mousse, la solution ne doit pas être injectée directement dans la poudre.
Une fois l'ajout terminé, le flacon doit être maintenu en position verticale pendant au moins 5 minutes afin de permettre la mouillabilité adéquate de la poudre lyophilisée. Ensuite, tourner délicatement et/ou retourner le flacon lentement pendant au moins 2 minutes jusqu'à la remise en dispersion complète de toute la poudre lyophilisée. Eviter toute formation de mousse. En cas de formation de mousse ou d'agrégats, maintenir le flacon en position verticale pendant au moins 15 minutes jusqu'à leur disparition.
La dispersion reconstituée doit avoir un aspect laiteux et homogène, sans précipité visible. Des agrégats de dispersion reconstituée peuvent se former. En cas de présence de précipités ou d'agrégats, le flacon doit être à nouveau délicatement retourné afin d'assurer la remise en dispersion complète avant utilisation.
Inspecter la dispersion contenue dans le flacon pour vérifier l'absence de toute particule. Ne pas administrer la dispersion reconstituée si des particules sont observées dans le flacon.
Le volume total exact de la dispersion à 5 mg/mL correspondant à la dose requise pour le patient doit être calculé et la quantité appropriée de BUGVI reconstitué doit être injectée dans une poche pour perfusion vide, stérile, en PVC ou non.
L'utilisation de dispositifs médicaux contenant de l'huile de silicone comme lubrifiant (c'est-à-dire, seringues et poches IV) pour reconstituer et administrer BUGVI peut entraîner la formation de filaments protéiques. Administrer BUGVI à l'aide d'un kit de perfusion muni d'un filtre de 15 μm, afin d'éviter l'administration de ces filaments. L'utilisation d'un filtre de 15 μm élimine les filaments et ne modifie pas les propriétés physiques ou chimiques du produit reconstitué.
L'utilisation de filtres dont la taille des pores est inférieure à 15 μm peut provoquer une obturation de ces filtres.
L'utilisation de poches ou de systèmes de perfusion sans phtalate de di-2-éthylhexyle (DEHP) n'est pas nécessaire pour préparer ou administrer les perfusions de BUGVI.
Après l'administration, il est recommandé de rincer la tubulure avec une solution injectable de chlorure de sodium à 0,9 mg/mL (9 %) afin de garantir l'administration de la dose complète.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I
Médicament soumis à prescription hospitalière.
Médicament à prescription réservée aux spécialistes en cancérologie, hématologie, oncologie médicale.
Médicament nécessitant une surveillance particulière pendant le traitement.
Poudre pour dispersion pour perfusion.
La dispersion reconstituée a un pH compris entre 6 et 7,5 et une osmolalité comprise entre 300 et 360 mOsm/kg.
La poudre est de couleur blanche à jaune.
Flacon de 50 mL avec bouchon (caoutchouc bromobutyle), muni d'une capsule de type « flip-off » en aluminium, contenant 100 mg de paclitaxel sous forme de nanoparticules liées à l'albumine.
Conditionnement d'un flacon.
Paclitaxel sous forme de nanoparticules liées à l'albumine...................................................... 100 mg
Pour un flacon de poudre.
Après reconstitution, chaque mL de dispersion contient 5 mg de paclitaxel sous forme de nanoparticules liées à l'albumine.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Albumine humaine (contenant du caprylate de sodium et du N-acétyl-L-tryptophan).