
ALTUVOCT 250 UI, poudre et solvant pour solution injectable, boîte de 1 flacon de poudre seringue de solvant de 3 ml
Dernière révision : 09/12/2024
Taux de TVA : 2.1%
Laboratoire exploitant : SWEDISH ORPHAN BIOVITRUM (SOBI)
Source :

Traitement et prophylaxie des épisodes hémorragiques chez les patients atteints d'hémophilie A (déficit congénital en facteur VIII).
ALTUVOCT est indiqué dans toutes les tranches d'âge.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Hypersensibilité
Des réactions d'hypersensibilité de type allergique sont possibles avec ALTUVOCT. En cas de survenue de symptômes d'hypersensibilité, il faut conseiller aux patients d'interrompre immédiatement l'administration du médicament et de contacter leur médecin. Les patients doivent être informés des signes précoces de réactions d'hypersensibilité tels que : urticaire, urticaire généralisée, oppression thoracique, respiration sifflante, hypotension et anaphylaxie.
En cas d'état de choc, le traitement médical standard relatif à l'état de choc doit être instauré.
Inhibiteurs
La formation d'anticorps neutralisants (inhibiteurs) dirigés contre le facteur VIII est une complication connue de la prise en charge des personnes atteintes d'hémophilie A. Ces inhibiteurs sont habituellement des immunoglobulines IgG dirigées contre l'activité pro-coagulante du facteur VIII, qui sont mesurées en unités Bethesda (UB) par mL de plasma à l'aide du test de Bethesda modifié. Le risque d'apparition d'inhibiteurs est corrélé à la sévérité de la maladie, ainsi qu'à l'exposition au facteur VIII. Ce risque est maximal au cours des 50 premiers jours d'exposition mais persiste tout au long de la vie, à un niveau toutefois peu élevé.
Les implications cliniques du développement d'anticorps dépendront du titre de l'inhibiteur, le risque de réponse clinique insuffisante étant plus faible si les titres d'inhibiteurs sont bas que s'ils sont élevés.
D'une manière générale, tous les patients traités avec un facteur VIII de coagulation doivent faire l'objet d'une surveillance étroite au moyen d'examens cliniques et biologiques appropriés afin de détecter l'apparition éventuelle d'inhibiteurs. Si les niveaux d'activité plasmatique du facteur VIII attendus ne sont pas atteints, ou si les saignements ne sont pas contrôlés au moyen d'une dose appropriée, des tests devront être réalisés pour rechercher la présence d'inhibiteurs du facteur VIII. Chez les patients présentant des taux élevés d'inhibiteurs, le traitement par le facteur VIII peut ne pas être efficace et d'autres options thérapeutiques devront être envisagées. La prise en charge de ces patients doit être assurée sous la supervision de médecins expérimentés dans le traitement de l'hémophilie et la gestion des inhibiteurs du facteur VIII.
Tests de surveillance en laboratoire
Si un test chromogénique ou un test de coagulation en un temps avec le réactif Actin‑FS est utilisé, le résultat doit être divisé par 2,5 pour obtenir le niveau d'activité approximatif du facteur VIII chez le patient (voir rubrique Posologie et mode d'administration). Il convient de noter que ce facteur de conversion n'est qu'une estimation (rapport moyen test chromogénique/test de coagulation en un temps avec Actin-FSL : 2,53 ; ET : 1,54 ; Q1 : 1,98 ; Q3 : 2,96 ; N = 3 353).
Événements cardiovasculaires
Chez les patients présentant des facteurs de risque cardiovasculaires préexistants, le traitement substitutif par des produits contenant le facteur VIII peut augmenter le risque cardiovasculaire.
Complications liées au cathéter
Si l'utilisation d'un dispositif d'accès veineux central (DAVC) est requise, le risque de complications liées au DAVC, telles que des infections locales, une bactériémie et une thrombose sur cathéter, doit être pris en compte.
Population pédiatrique
Les mises en garde et précautions s'appliquent aussi bien aux adultes qu'aux enfants.
Résumé du profil de sécurité
Une hypersensibilité ou des réactions allergiques (se manifestant par des symptômes tels que : un angio-œdème, une sensation de brûlure et de piqûre au site d'injection, des frissons, des bouffées vasomotrices, une urticaire généralisée, des céphalées, une urticaire, une hypotension, une léthargie, des nausées, une agitation, une tachycardie, une oppression thoracique, des picotements, des vomissements, une respiration sifflante) ont été observées dans de rares cas et peuvent, dans certains cas, évoluer vers une anaphylaxie sévère (y compris un choc).
Des anticorps neutralisants (inhibiteurs) peuvent apparaître chez les patients atteints d'hémophilie A traités par le facteur VIII, y compris par ALTUVOCT (voir rubrique Propriétés pharmacodynamiques). La présence de ces inhibiteurs peut se manifester par une réponse clinique insuffisante. Il est alors recommandé de contacter un centre spécialisé en hémophilie.
Liste des effets indésirables
Le tableau 2 ci-dessous présente les effets indésirables selon la classification de systèmes d'organes MedDRA (classes de systèmes d'organes et termes préconisés). La fréquence des effets indésirables a été établie sur la base des études cliniques de phase 3 menées chez 277 patients préalablement traités (PPT) atteints d'hémophilie A sévère, dont 161 (58,2 %) adultes (18 ans et plus), 37 (13,4 %) adolescents (12 à < 18 ans) et 79 (28,5 %) enfants de moins de 12 ans.
Des effets indésirables (EI) (récapitulés dans le tableau 2) ont été signalés chez 111 (40,1 %) des 277 patients traités dans le cadre d'une prophylaxie de routine ou d'un traitement à la demande.
Les fréquences sont définies selon les critères suivants : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Au sein de chaque groupe de fréquence, les effets indésirables sont présentés par ordre de gravité décroissante.
Tableau 2 : Effets indésirables signalés avec ALTUVOCT au cours des études cliniques
|
Classe de systèmes d'organes |
Effets indésirables |
Fréquence |
|
Affections du système nerveux |
Céphalée1 |
Très fréquent |
|
Affections gastro-intestinales |
Vomissements |
Fréquent |
|
Affections de la peau et du tissu sous-cutané |
Eczéma |
Fréquent |
|
Éruption cutanée2 |
Fréquent |
|
|
Urticaire3 |
Fréquent |
|
|
Affections musculosquelettiques et du tissu conjonctif |
Arthralgie |
Très fréquent |
|
Douleur dans les extrémités |
Fréquent |
|
|
Dorsalgie |
Fréquent |
|
|
Troubles généraux et anomalies au site d'administration |
Fièvre |
Fréquent |
|
Réaction au site d'injection4 |
Peu fréquent |
1
Céphalée, y compris migraine.
2 Éruption cutanée, y compris éruption maculo-papuleuse.
3 Urticaire, y compris urticaire papuleuse.
4 Réaction au site d'injection, y compris hématome au site d'injection et dermatite au site d'injection.
2 Éruption cutanée, y compris éruption maculo-papuleuse.
3 Urticaire, y compris urticaire papuleuse.
4 Réaction au site d'injection, y compris hématome au site d'injection et dermatite au site d'injection.
Population pédiatrique
S'agissant des effets indésirables, aucune différence liée à l'âge n'a été observée entre les patients pédiatriques et les patients adultes.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
SURVEILLANCE du traitement :
- Taux plasmatique de FVIII en cas d'intervention chirurgicale majeure ou d'hémorragie engageant le pronostic vital.
- Clinique et biologique afin de détecter l'apparition éventuelle d'inhibiteurs.
Tests de surveillance en laboratoire
Si un test chromogénique ou un test de coagulation en un temps avec le réactif Actin‑FS est utilisé, le résultat doit être divisé par 2,5 pour obtenir le niveau d'activité approximatif du facteur VIII chez le patient. Il convient de noter que ce facteur de conversion n'est qu'une estimation (rapport moyen test chromogénique/test de coagulation en un temps avec Actin-FSL : 2,53 ; ET : 1,54 ; Q1 : 1,98 ; Q3 : 2,96 ; N = 3 353).
- Taux plasmatique de FVIII en cas d'intervention chirurgicale majeure ou d'hémorragie engageant le pronostic vital.
- Clinique et biologique afin de détecter l'apparition éventuelle d'inhibiteurs.
Tests de surveillance en laboratoire
Si un test chromogénique ou un test de coagulation en un temps avec le réactif Actin‑FS est utilisé, le résultat doit être divisé par 2,5 pour obtenir le niveau d'activité approximatif du facteur VIII chez le patient. Il convient de noter que ce facteur de conversion n'est qu'une estimation (rapport moyen test chromogénique/test de coagulation en un temps avec Actin-FSL : 2,53 ; ET : 1,54 ; Q1 : 1,98 ; Q3 : 2,96 ; N = 3 353).
INTERROMPRE L'INJECTION ET CONTACTER IMMÉDIATEMENT LE MÉDECIN en cas de symptômes d'une réaction anaphylactique :
- gonflement du visage ;
- éruption cutanée ;
- démangeaisons généralisées ;
- éruption urticarienne ;
- bouffées congestives ;
- mal de tête ;
- tension artérielle basse ;
- sensation de malaise général ;
- oppression dans la poitrine ;
- difficultés à respirer ;
- sensation de brûlure et de piqûre au site d'injection ;
- frissons ;
- nausée ;
- agitation et battements de coeur rapides ;
- sensation d'étourdissement ;
- perte de conscience.
CONTACTER LE MÉDECIN en cas de saignements persistants.
Le facteur VIII n'a fait l'objet d'aucune étude sur les fonctions de reproduction chez l'animal. En raison de la rareté de l'hémophilie A chez la femme, il n'y a pas de donnée disponible sur l'utilisation de facteur VIII lors de la grossesse ou de l'allaitement. Par conséquent, le facteur VIII ne doit être utilisé pendant la grossesse et l'allaitement qu'en cas de nécessité absolue.
Aucune interaction n'a été rapportée entre les produits contenant le facteur VIII de coagulation humain (ADNr) et d'autres médicaments.
Aucune étude d'interaction n'a été réalisée.
Le traitement doit se faire sous la surveillance d'un médecin expérimenté dans le traitement de l'hémophilie.
Après avoir reçu une formation adéquate à la technique d'injection (voir la rubrique Précautions particulières d’élimination et de manipulation et la notice), le patient peut s'auto-injecter ALTUVOCT, ou un aidant peut l'administrer au patient, si le médecin le juge approprié.
Surveillance thérapeutique
La réponse au facteur VIII peut varier d'un patient à l'autre, les demi-vies et les niveaux de récupération pouvant être différents. Un ajustement de la dose en fonction du poids corporel peut être nécessaire en cas d'insuffisance pondérale ou de surpoids. La surveillance des taux de facteur VIII en vue de l'ajustement de la dose n'est habituellement pas nécessaire au cours de la prophylaxie de routine. En cas d'intervention chirurgicale majeure ou d'hémorragie engageant le pronostic vital, un dosage du taux de facteur VIII est nécessaire afin de déterminer la dose à administrer et la fréquence des injections.
Lorsqu'un test de coagulation in vitro en un temps basé sur le temps de céphaline activé (TCA) est utilisé pour déterminer l'activité du facteur VIII dans les échantillons de sang des patients, les résultats concernant l'activité plasmatique du facteur VIII peuvent être significativement influencés par le type de réactif de TCA et par l'étalon de référence utilisés pour le test. Des écarts significatifs peuvent également être observés entre les résultats obtenus par un test de coagulation en un temps basé sur le TCA et par le test chromogénique selon la Pharmacopée européenne. Ce point est particulièrement important lors d'un changement de laboratoire et/ou de réactif utilisé pour le test.
Il est recommandé d'utiliser un test de coagulation en un temps validé afin de déterminer l'activité plasmatique du facteur VIII associée à ALTUVOCT. Tout au long du développement clinique, un test de coagulation en un temps basé sur le réactif Actin‑FSL a été utilisé.
D'après les données d'une analyse comparative des échantillons issus d'une étude clinique, les résultats obtenus à l'aide d'un test chromogénique doivent être divisés par 2,5 pour établir approximativement l'activité du facteur VIII chez le patient (voir rubrique Mises en garde spéciales et précautions d'emploi). Par ailleurs, une étude de terrain comparant différents réactifs de TCA a indiqué que les niveaux d'activité du facteur VIII étaient environ 2,5 fois plus élevés lorsque le réactif Actin‑FS était utilisé à la place du réactif Actin‑FSL lors du test de coagulation en un temps, et que les résultats étaient environ 30 % inférieurs lorsque le réactif SynthASil était utilisé.
Posologie
La dose et la durée du traitement substitutif dépendent de la sévérité du déficit en facteur VIII, de la localisation et de l'intensité de l'épisode hémorragique, ainsi que de l'état clinique du patient.
Le nombre d'unités de facteur VIII administrées est exprimé en unités internationales (UI), conformément au standard actuel de l'OMS pour les produits contenant du facteur VIII. L'activité coagulante du facteur VIII dans le plasma est exprimée soit en pourcentage (par rapport au plasma humain normal), soit préférentiellement en unités internationales (par rapport au Standard International du facteur VIII plasmatique).
Une UI d'activité du facteur VIII correspond à la quantité de facteur VIII contenue dans un mL de plasma humain normal.
Pour la dose de 50 UI de facteur VIII par kg de poids corporel, la récupération plasmatique attendue in vivo du taux de facteur VIII, exprimée en UI/dL (ou en % de la normale), est estimée à l'aide de la formule suivante :
Augmentation estimée du facteur VIII (UI/dL ou % de la normale) = 50 UI/kg × 2 (UI/dL par UI/kg)
Traitement à la demande
Les doses d'ALTUVOCT à utiliser pour le traitement à la demande, le contrôle des épisodes hémorragiques et la gestion péri-opératoire sont indiquées dans le tableau 1.
Tableau 1 : Guide pour établir la posologie d'ALTUVOCT lors d'épisodes hémorragiques et de chirurgies
|
Degré d'hémorragie / Type d'intervention chirurgicale |
Dose recommandée |
Informations complémentaires |
|
Hémorragie Début d'hémarthrose, de saignement musculaire ou buccal |
Dose unique de 50 UI/kg |
En cas d'épisodes hémorragiques mineurs et modérés survenant dans les 2 à 3 jours suivant l'administration d'une dose prophylactique, une dose réduite de 30 UI/kg peut être utilisée. Une dose supplémentaire de 30 ou 50 UI/kg peut être envisagée au bout de 2 à 3 jours. |
|
Hémarthrose plus étendue, hémorragie musculaire ou hématome |
Dose unique de 50 UI/kg |
Des doses supplémentaires de 30 ou 50 UI/kg peuvent être envisagées tous les 2 à 3 jours jusqu'à résolution de l'hémorragie. |
|
Hémorragie engageant le pronostic vital |
Dose unique de 50 UI/kg |
Des doses supplémentaires de 30 ou 50 UI/kg peuvent être administrées tous les 2 à 3 jours jusqu'à disparition du risque vital. |
|
Chirurgie Chirurgie mineure, dont extraction dentaire |
Dose unique de 50 UI/kg |
Une dose supplémentaire de 30 ou 50 UI/kg peut être envisagée au bout de 2 à 3 jours. |
|
Chirurgie majeure |
Dose unique de 50 UI/kg |
Des doses supplémentaires de 30 ou 50 UI/kg peuvent être administrées selon les besoins cliniques tous les 2 à 3 jours jusqu'à cicatrisation satisfaisante de la plaie. |
Pour la reprise de la prophylaxie (le cas échéant) après le traitement d'une hémorragie, il est recommandé de respecter un intervalle d'au moins 72 heures entre la dernière dose de 50 UI/kg pour le traitement de l'hémorragie et la reprise de la dose prophylactique. Le traitement prophylactique peut ensuite être poursuivi normalement à la posologie habituellement utilisée chez le patient.
Prophylaxie
La dose recommandée pour la prophylaxie de routine chez l'adulte et l'enfant est de 50 UI/kg d'ALTUVOCT une fois par semaine.
Populations particulières
Population gériatrique
L'expérience chez les patients âgés de ≥ 65 ans est limitée. Les posologies recommandées sont les mêmes que chez les patients âgés de < 65 ans.
Population pédiatrique
Les posologies recommandées sont les mêmes que chez l'adulte.
Mode d'administration
Voie intraveineuse.
La dose complète d'ALTUVOCT doit être injectée par voie intraveineuse pendant 1 à 10 minutes, en fonction du niveau de confort du patient.
Pour les instructions concernant la dilution du médicament avant administration, voir la rubrique Précautions particulières d’élimination et de manipulation.
Durée de conservation :
Flacon non ouvert
4 ans
Pendant la durée de conservation, le médicament peut être conservé à température ambiante (jusqu'à 30 °C) pendant une période unique ne dépassant pas 6 mois. La date à laquelle le médicament est sorti du réfrigérateur doit être inscrite sur la boîte. Après avoir été conservé à température ambiante, le médicament ne doit pas être remis au réfrigérateur. Ne pas l'utiliser après la date de péremption imprimée sur le flacon ou plus de six mois après avoir sorti la boîte du réfrigérateur.
Après reconstitution
Le médicament doit être utilisé immédiatement après reconstitution. S'il n'est pas utilisé immédiatement, les durées et conditions de conservation avant utilisation relèvent de la responsabilité de l'utilisateur.
Précautions particulières de conservation :
À conserver au réfrigérateur (entre 2 °C et 8 °C).
Ne
pas congeler.
Conserver le flacon dans l'emballage extérieur, à l'abri de
la lumière.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique Durée de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Seuls l'adaptateur et le kit de perfusion fournis doivent être utilisés car le traitement pourrait échouer en raison de l'adsorption du facteur VIII de coagulation sur les surfaces internes de certains équipements d'injection.
Aucun symptôme de surdosage n'a été rapporté avec le facteur VIII de coagulation humain (ADNr).
Classe pharmacothérapeutique : antihémorragiques, facteur VIII de coagulation sanguine, Code ATC : B02BD02.
Mécanisme d'action
L'efanesoctocog alfa est un traitement de substitution du facteur VIII. Le facteur VIII activé agit comme un cofacteur vis-à-vis du facteur IX activé, accélérant la conversion du facteur X en facteur X activé. Le facteur X activé convertit la prothrombine en thrombine. La thrombine convertit alors le fibrinogène en fibrine, conduisant à la formation d'un caillot. L'hémophilie A est une maladie héréditaire de la coagulation sanguine liée à l'X, qui est due à une diminution du taux de facteur VIII:C fonctionnel et se traduit par des hémorragies au niveau des articulations, des muscles ou des organes internes, lesquelles surviennent spontanément ou à la suite d'un traumatisme accidentel ou chirurgical. Le traitement de substitution augmente le taux plasmatique de facteur VIII, ce qui permet une correction temporaire du déficit et de la tendance hémorragique.
Il est à noter que le taux de saignement annualisé (Annualized Bleeding Rate, ABR) n'est pas comparable entre les différents concentrés de facteur, ni entre les différentes études cliniques.
ALTUVOCT (efanesoctocog alfa), ou facteur VIII de coagulation recombinant fusionné au fragment Fc-facteur Von Willebrand (VWF)-protéine de fusion XTEN, est une protéine de fusion recombinante qui permet de remplacer temporairement le facteur VIII de coagulation manquant, qui est nécessaire à une hémostase efficace.
L'efanesoctocog alfa est une protéine de FVIII conçue pour ne pas se lier au VWF endogène, de façon à s'affranchir de la limite de demi-vie imposée par les interactions FVIII-VWF. Le domaine D'D3 du VWF est la région qui interagit avec le FVIII. L'ajout du domaine D'D3 du VWF à la protéine de fusion rFVIII-Fc assure la protection et la stabilité du FVIII et évite les interactions entre le FVIII et le VWF endogène, ce qui permet de s'affranchir de la limitation de la demi-vie du FVIII imposée par la clairance du VWF.
La région Fc de l'immunoglobuline G1 humaine (IgG1) se lie
au récepteur Fc néonatal (FcRn). Le FcRn fait partie d'une voie naturelle qui retarde
la dégradation lysosomale des immunoglobulines en les remettant en circulation
(recyclage) et en prolongeant ainsi la demi-vie plasmatique de la protéine de
fusion.
L'efanesoctocog alfa contient 2 polypeptides XTEN, qui
augmentent davantage sa pharmacocinétique (PK). Le domaine FVIII B naturel (à
l'exclusion de 5 acides aminés) est remplacé par le premier polypeptide XTEN,
inséré entre les résidus d'acides aminés FVIII N745 et E1649 ; le second XTEN
est inséré entre le domaine D'D3 et le fragment Fc.
Efficacité et sécurité cliniques
La sécurité, l'efficacité et la pharmacocinétique d'ALTUVOCT ont été évaluées dans le cadre de deux études cliniques de phase III multicentriques, prospectives, en ouvert (une étude menée chez des adultes et des adolescents [XTEND-1] et une étude pédiatrique menée chez des enfants âgés de < 12 ans [XTEND-Kids ; voir Population pédiatrique]) chez des patients préalablement traités (PPT) atteints d'hémophilie A sévère (activité du FVIII endogène < 1 % ou mutation génétique documentée concordant avec une hémophilie A sévère). La sécurité et l'efficacité à long terme d'ALTUVOCT sont également en cours d'évaluation dans le cadre d'une étude d'extension à long terme.
Toutes les études ont évalué l'efficacité de la prophylaxie de routine à la dose hebdomadaire de 50 UI/kg et ont établi l'efficacité hémostatique du traitement pour le contrôle des épisodes hémorragiques ainsi que la prise en charge péri-opératoire des patients lors des interventions chirurgicales majeures ou mineures. Par ailleurs, l'efficacité du traitement prophylactique par ALTUVOCT a également été comparée à celle du précédent facteur VIII prophylactique dans le cadre d'une comparaison intra-individuelle chez des patients ayant participé à une étude observationnelle prospective (OBS16221) avant leur inclusion dans l'étude XTEND-1.
Efficacité clinique de la prophylaxie de routine chez les adultes/adolescents
Dans l'étude conduite chez les adultes et les adolescents (XTEND-1) et clôturée, un total de 159 PPT (158 de sexe masculin et 1 de sexe féminin) atteints d'hémophilie A sévère ont été inclus. Les patients étaient âgés de 12 à 72 ans, dont 25 adolescents âgés de 12 à 17 ans. Les 159 patients inclus ont tous reçu au moins une dose d'ALTUVOCT et étaient évaluables en termes d'efficacité. Au total, 149 patients (93,7 %) ont terminé l'étude.
L'efficacité de la dose hebdomadaire de 50 UI/kg d'ALTUVOCT pour la prophylaxie de routine a été évaluée d'après l'estimation du taux de saignement annualisé (ABR) moyen (tableau 3) et en comparant l'ABR obtenu avec la prophylaxie de l'étude à l'ABR obtenu avec le facteur VIII prophylactique reçu avant l'étude (tableau 4). Au total, 133 adultes et adolescents, qui recevaient un facteur VIII prophylactique avant leur inclusion dans l'étude, ont été inclus dans le groupe traité par ALTUVOCT pour une prophylaxie de routine à la dose de 50 UI/kg une fois par semaine pendant 52 semaines (groupe A). En outre, 26 patients, qui recevaient un traitement par facteur VIII épisodique (à la demande) avant l'étude, ont reçu un traitement épisodique (à la demande) par ALTUVOCT à la dose de 50 UI/kg pendant 26 semaines, suivi d'une prophylaxie de routine à la dose de 50 UI/kg une fois par semaine pendant 26 semaines (groupe B). Globalement, 115 patients ont bénéficié d'un total d'au moins 50 jours d'exposition dans le groupe A et 17 patients ont reçu la prophylaxie de routine avec au moins 25 jours d'exposition dans le groupe B.
Tableau 3 : Résumé des taux de saignement annualisés (ABR) obtenus avec la prophylaxie par ALTUVOCT, le traitement à la demande par ALTUVOCT et après passage à la prophylaxie par ALTUVOCT chez les patients âgés de ≥ 12 ans
|
Critère d'évaluation[1] |
Groupe A Prophylaxie2 |
Groupe B À la demande[2] |
Groupe B Prophylaxie3 |
|
|
N = 133 |
N = 26 |
N = 26 |
|
Saignements |
|
|
|
|
ABR moyen (IC à 95 %)[3] |
0,71 (0,52 ; 0,97) |
21,41 (18,81 ; 24,37) |
0,70 (0,33 ; 1,49) |
|
ABR médian (EIQ) |
0,00 (0,00 ; 1,04) |
21,13 (15,12 ; 27,13) |
0,00 (0,00 ; 0,00) |
|
Patients sans aucun saignement, % |
64,7 |
0 |
76,9 |
|
Saignements spontanés |
|
|
|
|
ABR moyen (IC à 95 %)4 |
0,27 (0,18 ; 0,41) |
15,83 (12,27 ; 20,43) |
0,44 (0,16 ; 1,20) |
|
ABR médian (EIQ) |
0,00 (0,00 ; 0,00) |
16,69 (8,64 ; 23,76) |
0,00 (0,00 ; 0,00) |
|
Patients sans aucun saignement, % |
80,5 |
3,8 |
84,6 |
|
Saignements articulaires |
|
|
|
|
ABR moyen (IC à 95 %)4 |
0,51 (0,36 ; 0,72) |
17,48 (14,88 ; 20,54) |
0,62 (0,25 ; 1,52) |
|
ABR médian (EIQ) |
0,00 (0,00 ; 1,02) |
18,42 (10,80 ; 23,90) |
0,00 (0,00 ; 0,00) |
|
Patients sans aucun saignement, % |
72,2 |
0 |
80,8 |
La comparaison intra-individuelle des ABR entre la prophylaxie de l'étude et celle reçue avant l'étude a montré une réduction statistiquement significative de 77 % de l'ABR au cours de la prophylaxie de routine par ALTUVOCT par comparaison à la prophylaxie avec le facteur VIII reçu avant l'étude (voir le tableau 4).
Tableau 4 : Comparaison intra-individuelle des taux de saignement annualisés (ABR) entre le traitement prophylactique par ALTUVOCT et par facteur VIII reçu avant l'étude chez les patients âgés de ≥ 12 ans
|
Critère d'évaluation |
Prophylaxie de l'étude par ALTUVOCT 50 UI/kg 1×/sem. (N = 78) |
Prophylaxie par facteur VIII reçue avant l'étude2 (N = 78) |
|
Durée médiane d'observation (semaines) (EIQ) |
50,09 (49,07 ; 51,18) |
50,15 (43,86 ; 52,10) |
|
Saignements |
|
|
|
ABR moyen (IC à 95 %)1 |
0,69 (0,43 ; 1,11) |
2,96 (2,00 ; 4,37) |
|
Réduction de l'ABR, % (IC à 95 %) Valeur de p |
77 (58 ; 87) < 0,0001 |
|
|
Patients sans aucun saignement, % |
64,1 |
42,3 |
|
ABR médian (EIQ) |
0,00 (0,00 ; 1,04) |
1,06 (0,00 ; 3,74) |
1
Basé sur un modèle binomial négatif.
2 Étude observationnelle prospective (OBS16221).
ABR = taux de saignement annualisé ; EIQ = écart interquartile, entre le 25e et le 75e centile ; IC = intervalle de confiance.
2 Étude observationnelle prospective (OBS16221).
ABR = taux de saignement annualisé ; EIQ = écart interquartile, entre le 25e et le 75e centile ; IC = intervalle de confiance.
La comparaison intra-individuelle (N = 26) des ABR entre les 26 premières semaines de traitement à la demande par ALTUVOCT et les 26 semaines suivantes en prophylaxie hebdomadaire par ALTUVOCT (groupe B) a montré une réduction cliniquement importante des saignements, à hauteur de 97 %, avec le traitement prophylactique hebdomadaire et une augmentation de la proportion de patients sans aucun saignement, passée de 0 à 76,9 %.
Efficacité pour le contrôle des saignements
Au cours de l'étude menée chez les adultes et les adolescents (XTEND-1), 362 épisodes hémorragiques au total ont été traités avec ALTUVOCT, la plupart étant survenus pendant le traitement à la demande dans le groupe B. La majorité des épisodes hémorragiques étaient localisés au niveau des articulations. La réponse à la première injection a été évaluée par les patients au moins 8 heures après le traitement. Une échelle à 4 points (réponse excellente, bonne, moyenne et aucune réponse) a été utilisée pour évaluer la réponse. L'efficacité pour le contrôle des épisodes hémorragiques chez les patients âgés de ≥ 12 ans est résumée dans le tableau 5. Le contrôle des épisodes hémorragiques a été similaire entre les groupes de traitement.
Tableau 5 : Résumé de l'efficacité pour le contrôle des saignements chez les patients âgés de ≥ 12 ans
|
Nombre d'épisodes hémorragiques |
|
(N = 362) |
|
Nombre d'injections pour traiter l'épisode hémorragique, N (%) |
1 injection 2 injections > 2 injections | 350 (96,7)
11 (3,0) 1 (0,3) |
|
Dose totale médiane pour traiter un épisode hémorragique (UI/kg) (EIQ) |
|
50,93 (50,00 ; 51,85) |
|
Nombre d'injections évaluables |
|
(N = 332) |
|
Réponse au traitement de l'épisode hémorragique, N (%) |
Excellente ou bonne Moyenne Aucune réponse |
315 (94,9) 14 (4,2) 3 (0,9) |
Prise en charge péri-opératoire des saignements
L'hémostase péri-opératoire a été évaluée dans le cadre de 49 interventions chirurgicales majeures chez 41 patients (32 adultes et 9 adolescents et enfants) au cours des différentes études de phase III. Sur les 49 interventions chirurgicales majeures, 48 ont nécessité une seule dose préopératoire pour maintenir l'hémostase au cours de l'intervention ; pour 1 intervention chirurgicale majeure au cours de la prophylaxie de routine, aucune dose de charge préopératoire n'a été administrée le jour ou la veille de l'intervention. La dose médiane par injection préopératoire était de 50 UI/kg (intervalle : 12,7 - 84,7). La consommation totale moyenne (ET) et le nombre d'injections reçues au cours de la période péri-opératoire (depuis la veille de l'intervention jusqu'au jour 14 après l'intervention) ont été respectivement de 171,85 (51,97) UI/kg et de 3,9 (1,4).
L'évaluation clinique de la réponse hémostatique au cours de l'intervention chirurgicale majeure a été réalisée à l'aide d'une échelle à 4 points (excellente, bonne, moyenne ou mauvaise/nulle). L'effet hémostatique d'ALTUVOCT a été jugé « excellent » ou « bon » pour 48 des 49 interventions chirurgicales (98 %). Aucune intervention chirurgicale n'a donné lieu à une réponse jugée « mauvaise/nulle » et il n'y avait aucune évaluation « manquante ».
Les interventions chirurgicales majeures évaluées comprenaient des interventions orthopédiques majeures telles que des arthroplasties (remplacement des articulations du genou, de la hanche et du coude), des révisions articulaires et des procédures de fusion de la cheville. Les autres interventions chirurgicales majeures comprenaient l'extraction de molaires, la restauration et l'extraction dentaire, la circoncision, la résection d'une malformation vasculaire, la réparation d'une hernie et la rhinoplastie/génioplastie. De plus, 25 interventions chirurgicales mineures ont été évaluées ; l'hémostase a été jugée « excellente » dans tous les cas disponibles pour l'évaluation.
Immunogénicité
L'immunogénicité a été évaluée au cours des études cliniques menées avec ALTUVOCT chez des adultes et des enfants préalablement traités atteints d'hémophilie A sévère. Aucun développement d'inhibiteurs d'ALTUVOCT n'a été détecté durant les études cliniques.
Au cours des études cliniques de phase III (durée médiane de traitement de 96,3 semaines), 4 patients évaluables sur 276 (1,4 %) ont développé des anticorps anti-médicament (AAM) transitoires apparus sous traitement. Aucun signe d'un impact des AAM sur la pharmacocinétique, l'efficacité ou la sécurité du médicament n'a été observé.
Population pédiatrique
Prophylaxie de routine
L'efficacité de la prophylaxie de routine hebdomadaire par ALTUVOCT à 50 UI/kg chez les enfants âgés de < 12 ans a été évaluée d'après l'estimation de l'ABR moyen. Au total, 74 enfants (38 enfants de < 6 ans et 36 enfants de 6 à < 12 ans) ont été inclus en vue de recevoir une prophylaxie de routine par ALTUVOCT à la dose de 50 UI/kg par voie intraveineuse une fois par semaine pendant 52 semaines. Chez la totalité des 74 patients, la prophylaxie de routine a abouti à un ABR moyen global (IC à 95 %) de 0,9 (0,6 ; 1,4) et à un ABR médian (Q1 ; Q3) de 0 (0 ; 1,0) pour les saignements traités.
Une analyse de sensibilité (N = 73), avec exclusion d'un patient qui n'avait pas reçu le traitement prophylactique hebdomadaire comme prévu dans le protocole pendant une période prolongée, a mis en évidence un ABR moyen (IC à 95 %) de 0,6 (0,4 ; 0,9) pour les saignements traités (médiane [Q1 ; Q3] de 0 [0 ; 1,0]. Chez 47 enfants (64,4 %), aucun épisode hémorragique nécessitant un traitement n'est survenu. L'ABR moyen (IC à 95 %) pour les saignements spontanés traités a été de 0,2 (0 ; 0,3) (médiane [Q1 ; Q3] de 0 [0 ; 0]). Pour les saignements articulaires traités, l'ABR moyen (IC à 95 %) a été de 0,3 (0,2 ; 0,6) et la médiane (Q1 ; Q3) a été de 0 (0 ; 0).
Contrôle des saignements
L'efficacité pour le contrôle des saignements chez les enfants âgés de < 12 ans a été évaluée dans le cadre de l'étude pédiatrique, en excluant un patient qui n'avait pas reçu le traitement prophylactique hebdomadaire comme prévu dans le protocole pendant une période prolongée. Au total, 43 épisodes hémorragiques ont été traités avec ALTUVOCT. Une résolution du saignement a été obtenue avec une seule injection de 50 UI/kg d'ALTUVOCT dans 95,3 % des cas d'épisodes hémorragiques. La dose totale médiane (Q1 ; Q3) utilisée pour traiter un épisode hémorragique était de 52,6 UI/kg (50,0 ; 55,8).
La pharmacocinétique (PK) d'ALTUVOCT a été évaluée au cours des études de phase III XTEND-1 et XTEND-Kids, menées respectivement chez 159 adultes et adolescents et chez 74 enfants âgés de < 12 ans, qui ont reçu des injections intraveineuses hebdomadaires de 50 UI/kg. Parmi les enfants âgés de < 12 ans, des profils PK d'ALTUVOCT à dose unique étaient disponibles chez 37 patients.
L'efanesoctocog alfa a présenté une demi-vie environ 4 fois plus longue que celle des produits à base de facteur VIII à demi-vie standard, et environ 2,5 à 3 fois plus longue que celle des produits à base de facteur VIII à demi-vie prolongée. Les paramètres PK mesurés après administration d'une dose unique d'ALTUVOCT sont présentés dans le tableau 6. Les paramètres PK ont été établis d'après l'activité plasmatique du facteur VIII mesurée par le test de coagulation en un temps basé sur le TCA. Après administration d'une dose unique de 50 UI/kg, ALTUVOCT a présenté une activité élevée et durable du facteur VIII, avec une demi-vie prolongée dans tous les groupes d'âge. Une tendance à l'augmentation de l'ASC et à la diminution de la clairance a été observée avec l'âge dans les cohortes pédiatriques. Le profil PK à l'état d'équilibre (semaine 26) était comparable au profil PK obtenu après la première dose.
Tableau 6 : Paramètres pharmacocinétiques après administration d'une dose unique d'ALTUVOCT selon l'âge (test de coagulation en un temps avec le réactif Actin‑FSL)
|
Paramètres PK Moyenne (ET) |
Étude pédiatrique |
Étude chez les adultes et adolescents |
||
|
|
1 à < 6 ans |
6 à < 12 ans |
12 à < 18 ans |
Adultes |
|
N = 18 |
N = 18 |
N = 25 |
N = 134 |
|
|
ASC0-tau, UI*h/dL |
6 800 (1 120)b |
7 190 (1 450) |
8 350 (1 550) |
9 850 (2 010)a |
|
t½z, h |
38,0 (3,72) |
42,4 (3,70) |
44,6 (4,99) |
48,2 (9,31) |
|
CL, mL/h/kg |
0,742 (0,121) |
0,681 (0,139) |
0,582 (0,115) |
0,493 (0,121)a |
|
Véq, mL/kg |
36,6 (5,59) |
38,1 (6,80) |
34,9 (7,38) |
31,0 (7,32)a |
|
TRM, h |
49,6 (5,45) |
56,3 (5,10) |
60,0 (5,54) |
63,9 (10,2)a |
|
Cmax, UI/dL |
143 (57,8) |
113 (22,7) |
118 (24,9) |
133 (33,8) |
|
Récupération incrémentielle, UI/dL par UI/kg |
2,81 (1,1) |
2,24 (0,437) |
2,34 (0,490) |
2,64 (0,665) |
a Calcul basé sur 128 profils.
b N = 17
ASC0-tau = aire sous la courbe de l'activité en fonction du temps sur l'intervalle entre les doses ;
CL = clairance ; TRM = temps de résidence moyen ; ET = écart type ; t½z = demi-vie terminale ; Véq = volume de distribution à l'état d'équilibre ; Cmax = activité maximale
b N = 17
ASC0-tau = aire sous la courbe de l'activité en fonction du temps sur l'intervalle entre les doses ;
CL = clairance ; TRM = temps de résidence moyen ; ET = écart type ; t½z = demi-vie terminale ; Véq = volume de distribution à l'état d'équilibre ; Cmax = activité maximale
Dans l'étude XTEND-1, à l'état d'équilibre, l'activité du facteur VIII s'est maintenue à un niveau normal ou proche de la normale (> 40 UI/dL) pendant une durée moyenne (ET) de 4,1 (0,7) jours avec le traitement prophylactique par ALTUVOCT administré une fois par semaine chez les adultes. Une activité du facteur VIII supérieure à 10 UI/dL s'est maintenue chez 83,5 % des adultes et adolescents tout au long de l'étude. Chez les enfants âgés de < 12 ans, avec le traitement hebdomadaire par ALTUVOCT à l'état d'équilibre, l'activité du facteur VIII s'est maintenue à un niveau normal ou proche de la normale (> 40 UI/dL) pendant 2 à 3 jours et à un niveau > 10 UI/dL pendant environ 7 jours (voir le tableau 7).
Tableau 7 : Paramètres pharmacocinétiques d'ALTUVOCT à l'état d'équilibre selon l'âge (test de coagulation en un temps avec le réactif Actin‑FSL)
|
Paramètres PK Moyenne (ET) |
Étude pédiatriquea |
Étude chez les adultes et adolescentsa |
||
|
|
1 à < 6 ans |
6 à < 12 ans |
12 à < 18 ans |
Adultes |
|
N = 37 |
N = 36 |
N = 24 |
N = 125 |
|
|
Pic d'activité, UI/dL |
136 (48,9) (N = 35) |
131 (36,1) (N = 35) |
124 (31,2) |
150 (35,0) (N = 124) |
|
Récupération incrémentielle, UI/dL par UI/kg |
2,22 (0,83) (N = 35) |
2,10 (0,73) (N = 35) |
2,25 (0,61) (N = 22) |
2,64 (0,61) (N = 120) |
|
Délai d'obtention de 40 UI/dL, h |
68,0 (10,5)b |
80,6 (12,3)b |
81,5 (12,1)c |
98,1 (20,1)c |
|
Délai d'obtention de 20 UI/dL, h |
109 (14,0)b |
127 (14,5)b |
130 (15,7)c |
150 (27,7)c |
|
Délai d'obtention de 10 UI/dL, h |
150 (18,2)b |
173 (17,1)b |
179 (20,2)c |
201 (35,7)c |
|
Activité minimale, UI/dL |
10,9 (19,7) (N = 36) |
16,5 (23,7) |
9,23 (4,77) (N = 22) |
18,0 (16,6) (N = 123) |
a Le pic d'activité, l'activité minimale et la
récupération incrémentielle à l'état d'équilibre ont été calculés en utilisant
les mesures disponibles à la semaine 52/lors de la visite de prélèvement PK en
fin d'étude.
b Le délai d'obtention des différents niveaux d'activité du facteur VIII a été prédit à l'aide d'un modèle de pharmacocinétique de population pour les patients pédiatriques.
c Le délai d'obtention des différents niveaux d'activité du facteur VIII a été prédit à l'aide d'un modèle de pharmacocinétique de population pour les patients adultes.
Pic d'activité = 15 min après la dose à l'état d'équilibre ; activité minimale = valeur de l'activité du facteur VIII avant la dose à l'état d'équilibre ; ET = écart type
b Le délai d'obtention des différents niveaux d'activité du facteur VIII a été prédit à l'aide d'un modèle de pharmacocinétique de population pour les patients pédiatriques.
c Le délai d'obtention des différents niveaux d'activité du facteur VIII a été prédit à l'aide d'un modèle de pharmacocinétique de population pour les patients adultes.
Pic d'activité = 15 min après la dose à l'état d'équilibre ; activité minimale = valeur de l'activité du facteur VIII avant la dose à l'état d'équilibre ; ET = écart type
ALTUVOCT n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Les données non cliniques issues des études de toxicologie en administration répétée chez le rat et le singe (incluant des mesures de pharmacologie de sécurité) et d'une étude d'hémocompatibilité in vitro n'ont pas révélé de risque particulier pour l'homme. Aucune étude n'a été menée pour évaluer la génotoxicité, la cancérogenèse, la toxicité sur les fonctions de reproduction ou le développement embryo-fœtal.
ALTUVOCT doit être administré par voie intraveineuse après reconstitution de la poudre à l'aide du solvant fourni dans la seringue. Le flacon doit être remué délicatement avec un mouvement circulaire jusqu'à dissolution complète de la poudre. Après reconstitution, la solution doit être limpide et incolore ou légèrement opalescente. La solution ne doit pas être utilisée si elle est trouble ou présente des dépôts.
Les règles d'asepsie doivent toujours être respectées.
Informations complémentaires concernant la reconstitution
ALTUVOCT doit être administré par injection intraveineuse après dissolution de la poudre pour solution injectable à l'aide du solvant fourni dans la seringue préremplie. La boîte d'ALTUVOCT contient :
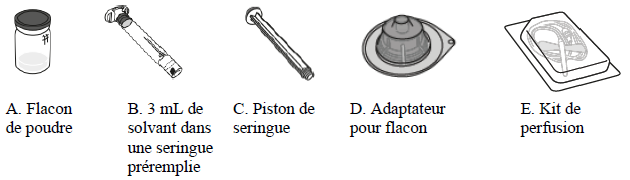
Vous aurez également besoin de tampons stériles imprégnés d'alcool (F). Ceux-ci ne sont pas fournis dans la boîte d'ALTUVOCT.
Une grande seringue (G) qui vous aura été remise séparément peut être utilisée pour aspirer dans une seule et même seringue le contenu de plusieurs flacons. Si vous ne disposez pas d'une grande seringue, suivez les étapes 6 à 8 pour administrer la solution contenue dans chaque seringue.
ALTUVOCT ne doit pas être mélangé avec d'autres solutions injectables ou pour perfusion.
Lavez-vous les mains avant d'ouvrir l'emballage.
Reconstitution
|
1. Préparez le flacon |
|
|
a. Retirez l'opercule du flacon Tenez le flacon de poudre (A) sur une surface plane et propre et retirez l'opercule en plastique. |
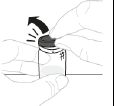 |
|
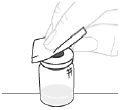
b. Nettoyez le haut du flacon Nettoyez le haut du flacon à l'aide d'un tampon imprégné d'alcool. Veillez à ne rien mettre en contact avec le haut du flacon après l'avoir nettoyé.
|
 |
|
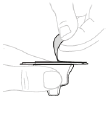
c. Ouvrez l'emballage de l'adaptateur pour flacon Ouvrez l'emballage de l'adaptateur pour flacon en retirant l'opercule de protection en papier (D). Ne touchez pas l'adaptateur pour flacon et ne le sortez pas de son emballage.
|
 |
|
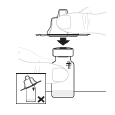
d. Fixez l'adaptateur pour flacon Placez l'adaptateur pour flacon directement sur le haut du flacon, en le laissant toujours dans son emballage. Appuyez fermement vers le bas jusqu'à ce que l'adaptateur s'enclenche. Le perforateur pénètre alors au travers du bouchon du flacon.
|
 |
|
2. Préparez la seringue
|
|
|
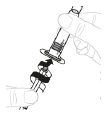
a. Fixez le piston de seringue Insérez le piston (C) dans la seringue de 3 mL (B). Faites tourner le piston dans le sens des aiguilles d'une montre jusqu'à ce qu'il soit solidement calé.
|
 |
|
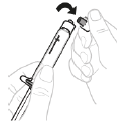
b. Retirez le bouchon de la seringue Détachez la partie supérieure du bouchon blanc de la seringue de 3 mL au niveau des perforations et mettez-la de côté.
|
 |
|
3. Fixez la seringue sur le flacon |
|
|
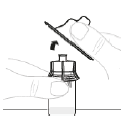
a. Retirez l'emballage de l'adaptateur pour flacon Retirez l'emballage de l'adaptateur pour flacon et jetez-le.
|
 |
|
b. Fixez la seringue sur l'adaptateur pour flacon Tenez l'adaptateur pour flacon par son extrémité inférieure. Placez l'extrémité de la seringue sur le haut de l'adaptateur pour flacon. Faites tourner la seringue dans le sens des aiguilles d'une montre pour la fixer solidement.
|
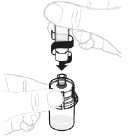 |
|
4. Dissolvez la poudre avec le solvant
|
|
|
a.Transférez le solvant dans le flacon Appuyez lentement sur le piston afin d'injecter la totalité du solvant dans le flacon.
|
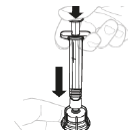 |
|
b. Dissolvez la poudre En gardant le pouce sur le piston, remuez délicatement le flacon en le faisant tourner jusqu'à dissolution de la poudre. Ne le secouez pas.
|
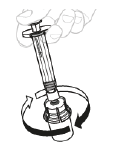 |
|
c. Inspectez la solution Inspectez la solution avant administration. Elle doit être limpide et incolore. N'utilisez pas la solution si elle est trouble ou contient des particules visibles.
|
|
|
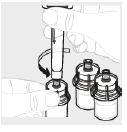
5. En cas d'utilisation de plusieurs flacons |
|
|
Si vous avez besoin de plusieurs flacons pour votre dose, suivez les étapes ci-dessous (5a et 5b) ; sinon, passez à l'étape 6. |
|
|
a. Répétez les étapes 1 à 4 Répétez les étapes 1 à 4 avec tous les flacons jusqu'à ce que vous ayez préparé une quantité suffisante de solution pour votre dose. Retirez les seringues de 3 mL de chaque flacon (voir l'étape 6b), en laissant la solution dans chacun des flacons.
|
 |
|
b. Utilisation d'une grande seringue (G) Pour chaque flacon, fixez la grande seringue (G) sur l'adaptateur pour flacon (voir l'étape 3b) et passez à l'étape 6 afin de rassembler la solution provenant de chaque flacon dans la grande seringue. Si vous n'avez besoin que d'une partie du contenu d'un flacon, utilisez la graduation de la seringue pour voir la quantité de solution aspirée.
|
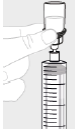 |
|
6. Aspirez la solution dans la seringue
|
|
|
a. Aspirez la solution Tenez la seringue pointée vers le haut. Tirez lentement sur le piston afin d'aspirer toute la solution dans la seringue.
|
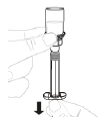 |
| b. Détachez la seringue Retirez la seringue du flacon en tenant l'adaptateur pour flacon. Faites tourner la seringue dans le sens inverse des aiguilles d'une montre pour la détacher. |
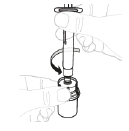 |
 Ne touchez pas l'intérieur du bouchon ni l'extrémité de la seringue.
Ne touchez pas l'intérieur du bouchon ni l'extrémité de la seringue. Il est recommandé d'utiliser ALTUVOCT immédiatement après reconstitution (voir la rubrique Durée de conservation).
Administration
|
7. Préparez l'injection |
|
|
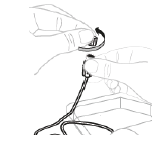
a. Retirez le capuchon de la tubulure Ouvrez l'emballage du kit de perfusion (E) (n'utilisez pas le kit s'il est endommagé). Retirez le capuchon de la tubulure.
|
 |
|
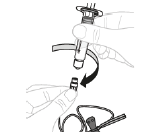
b. Raccordez la seringue Raccordez la seringue préparée à l'extrémité de la tubulure du kit de perfusion en la faisant tourner dans le sens des aiguilles d'une montre. |
 |
|
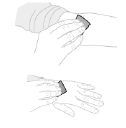
c. Préparez le site de perfusion Si nécessaire, posez un garrot. Nettoyez le site d'injection à l'aide d'un tampon imprégné d'alcool (F). |
 |
|
d. Éliminez l'air de la seringue et de la tubulure Éliminez l'air en pointant la seringue vers le haut et en appuyant délicatement sur le piston. Ne faites pas sortir la solution de l'aiguille.
|
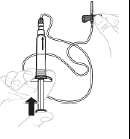 |
|
8. Injectez la solution |
|
|
a. Insérez l'aiguille Retirez le capuchon de protection de l'aiguille. Introduisez l'aiguille dans une veine et retirez le garrot, le cas échéant.
|
|
|
b. Injectez la solution La solution préparée doit être injectée par voie intraveineuse pendant 1 à 10 minutes, en fonction du niveau de confort du patient. |
|
|
9. Jetez le matériel de manière sécurisée |
|
|
a. Retirez l'aiguille Retirez l'aiguille. Pliez le protège-aiguille ; il doit s'enclencher en position fermée. | 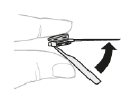 |
|
b. Élimination sécurisée Veillez à éliminer tous les composants utilisés dans le kit fourni (hormis l'emballage) de manière sécurisée dans une boîte de récupération des déchets médicaux.
|
|
 Vous
pouvez utiliser un pansement pour maintenir les ailettes en plastique de
l'aiguille en place au niveau du site d'injection et éviter que l'aiguille
bouge.
Vous
pouvez utiliser un pansement pour maintenir les ailettes en plastique de
l'aiguille en place au niveau du site d'injection et éviter que l'aiguille
bouge. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Prescription hospitalière.
Poudre et solvant pour solution injectable.
Poudre : poudre ou agglomérat lyophilisé, blanc à
blanc cassé.
Solvant : solution limpide et incolore.
pH : 6,5 à 7,2
Osmolalité : 586 à 688 mOsm/kg
Chaque boîte d'ALTUVOCT 250 UI poudre et solvant pour solution injectable contient :
- un flacon de verre (de type I) contenant la poudre, fermé par un bouchon en caoutchouc chlorobutyle ;
- un adaptateur pour flacon stérile pour la reconstitution ;
-
une seringue préremplie en verre contenant 3 mL de solvant, avec un
bouchon-piston en caoutchouc bromobutyle ;
- un piston de seringue ;
- un kit de perfusion stérile.
Chaque flacon contient nominalement 250 UI d'efanesoctocog alfa. ALTUVOCT contient environ 83 UI/mL de facteur VIII de coagulation humain, efanesoctocog alfa, après reconstitution.
L'activité (UI) est déterminée par la méthode du test de coagulation en un temps basé sur le temps de céphaline activé (TCA) en utilisant Actin-FSL comme réactif.
L'efanesoctocog alfa (facteur VIII de coagulation humain [ADNr]) est une protéine de 2 829 acides aminés.
L'efanesoctocog alfa est produit par la technologie de l'ADN recombinant dans une lignée de cellules rénales embryonnaires humaines (HEK). Aucune matière première d'origine humaine ou animale n'est utilisée au cours du processus de fabrication.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Poudre
Saccharose
Chlorure de calcium dihydraté (E 509)
Histidine
Chlorhydrate d'arginine
Polysorbate 80 (E 433)
Solvant
Eau pour préparations injectables