
OPDUALAG 240 mg-80 mg, solution à diluer pour perfusion, boîte de 1 flacon de 20 ml
Dernière révision : 12/12/2024
Taux de TVA : 2.1%
Laboratoire exploitant : BRISTOL-MYERS SQUIBB
Source :

Opdualag est indiqué en première ligne de traitement du mélanome avancé (non résécable ou métastatique) chez les adultes et les adolescents âgés de 12 ans et plus avec une expression de PD-L1 au niveau des cellules tumorales inférieure à 1 %.
Hypersensibilité aux substances actives ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Évaluation du statut PD-L1
Pour évaluer le statut PD-L1 de la tumeur, il est important d'employer une méthodologie fiable et dûment validée.
Effets indésirables d'origine immunologique
Des effets indésirables d'origine immunologique peuvent survenir en cas d'association du nivolumab avec le relatlimab, nécessitant une prise en charge appropriée, incluant l'instauration de corticoïdes et des modifications de traitement (voir rubrique Posologie et mode d'administration).
Des effets indésirables d'origine immunologique affectant plus d'un système d'organes peuvent survenir simultanément.
Les patients doivent être continuellement surveillés (au moins jusqu'à 5 mois après la dernière perfusion), puisqu'un effet indésirable avec Opdualag peut survenir à tout moment pendant ou après l'arrêt du traitement.
En cas de suspicion d'effets indésirables d'origine immunologique, une évaluation appropriée doit être effectuée afin de confirmer l'étiologie ou d'exclure d'autres causes. En fonction de la sévérité de l'effet indésirable, Opdualag doit être suspendu et des corticoïdes doivent être administrés. Si une immunosuppression par corticoïdes est utilisée pour traiter un effet indésirable, une décroissance progressive des doses sur une période d'au moins un mois doit être initiée à partir de l'amélioration. Une diminution rapide des doses peut entraîner une aggravation ou une récidive de l'effet indésirable. Des traitements immunosuppresseurs non corticostéroïdiens doivent être ajoutés en cas d'aggravation ou d'absence d'amélioration malgré l'utilisation de corticoïdes.
Opdualag ne doit pas être repris tant que le patient reçoit des doses immunosuppressives de corticoïdes ou d'autres médicaments immunosuppresseurs. Une prophylaxie antibiotique peut être utilisée pour prévenir les infections opportunistes chez les patients recevant des traitements immunosuppresseurs.
Opdualag doit être arrêté définitivement en cas d'effet indésirable sévère récurrent d'origine immunologique, et pour tout effet indésirable d'origine immunologique pouvant menacer le pronostic vital.
Pneumopathie inflammatoire d'origine immunologique
Des pneumopathies inflammatoires ou interstitielles diffuses sévères, dont un cas d'issue fatale, ont été observées avec le nivolumab associé au relatlimab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour déceler l'apparition de signes et symptômes de pneumopathie inflammatoire, tels que des modifications radiologiques (ex. : opacités focales en verre dépoli, infiltrats localisés), une dyspnée et une hypoxie. Les étiologies infectieuses et pathologiques doivent être éliminées.
En cas de pneumopathie inflammatoire de grade 3 ou 4, Opdualag doit être arrêté définitivement et une corticothérapie à la dose de 2 à 4 mg/kg/jour de méthylprednisolone ou équivalent doit être instaurée.
En cas de pneumopathie inflammatoire (symptomatique) de grade 2, Opdualag doit être suspendu et une corticothérapie à la dose de 1 mg/kg/jour de méthylprednisolone ou équivalent doit être instaurée. Après amélioration, Opdualag peut être repris après réduction progressive des corticoïdes. En cas d'aggravation ou d'absence d'amélioration malgré l'instauration d'une corticothérapie, les doses de corticoïdes doivent être augmentées à la dose de 2 à 4 mg/kg/jour de méthylprednisolone ou équivalent, et Opdualag doit être arrêté définitivement.
Colite d'origine immunologique
Des diarrhées ou des colites sévères ont été observées avec le nivolumab associé au relatlimab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour déceler l'apparition de diarrhées et d'autres symptômes de colites, tels que des douleurs abdominales et la présence de mucus et/ou de sang dans les selles. Une infection / réactivation du cytomégalovirus (CMV) a été rapportée chez des patients ayant des colites d'origine immunologique réfractaires aux corticoïdes. Les étiologies infectieuses et autres des diarrhées doivent être exclues, par conséquent, des analyses de laboratoire appropriées et des examens complémentaires doivent être effectués. Si le diagnostic de colite d'origine immunologique réfractaire aux corticoïdes est confirmé, l'ajout d'un agent immunosuppresseur alternatif au traitement par corticoïdes ou le remplacement du traitement par corticoïdes doivent être envisagés.
En cas de diarrhée ou de colite de grade 4, Opdualag doit être arrêté définitivement et une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit être instaurée.
Opdualag doit être suspendu en cas de diarrhée ou de colite de grade 3 et une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit être instaurée. Après amélioration, Opdualag peut être repris après réduction progressive des corticoïdes. En cas d'aggravation ou d'absence d'amélioration malgré l'instauration d'une corticothérapie, Opdualag doit être arrêté définitivement.
En cas de diarrhée ou de colite de grade 2, Opdualag doit être suspendu. Les diarrhées ou les colites persistantes doivent être traitées par une corticothérapie à la dose de 0,5 à 1 mg/kg/jour de méthylprednisolone ou équivalent. Après amélioration, Opdualag peut être repris après réduction progressive des corticoïdes, si nécessaire. En cas d'aggravation ou d'absence d'amélioration malgré l'instauration d'une corticothérapie, la dose de corticoïdes doit être augmentée à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent, et Opdualag doit être arrêté définitivement.
Hépatite d'origine immunologique
Des hépatites sévères ont été observées avec le nivolumab associé au relatlimab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour déceler l'apparition de signes et symptômes d'hépatite, tels que des augmentations des transaminases et de la bilirubine totale. Les étiologies infectieuses et pathologiques doivent être éliminées.
En cas d'augmentation des ASAT ou ALAT supérieure à 5 fois la LSN, indépendamment de la valeur de référence, d'augmentation de la bilirubine totale supérieure à 3 fois la LSN, d'augmentation concomitante des ASAT ou ALAT supérieure à 3 fois la LSN et de la bilirubine totale supérieure à 2 fois la LSN, Opdualag doit être arrêté définitivement et une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit être instaurée.
En cas d'augmentation des ASAT ou ALAT supérieure à 3 fois et jusqu'à 5 fois la LSN, ou en cas d'augmentation de la bilirubine totale supérieure à 1,5 fois et jusqu'à 3 fois la LSN, Opdualag doit être suspendu. Les augmentations persistantes des valeurs biologiques doivent être prises en charge par une corticothérapie à la dose de 0,5 à 1 mg/kg/jour de méthylprednisolone ou équivalent. Après amélioration, Opdualag peut être repris après réduction progressive des corticoïdes, si nécessaire. En cas d'aggravation ou d'absence d'amélioration malgré l'instauration d'une corticothérapie, la dose de corticoïdes doit être augmentée à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent, et Opdualag doit être arrêté définitivement.
Néphrite et dysfonction rénale d'origine immunologique
Des néphrites et des dysfonctions rénales sévères ont été observées avec le nivolumab associé au relatlimab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour déceler l'apparition de signes et symptômes de néphrite ou de dysfonction rénale. La plupart des patients ont présenté des augmentations asymptomatiques de la créatinine sanguine. Toute autre étiologie pathologique doit être écartée.
En cas d'élévation de grade 4 de la créatinine sanguine, Opdualag doit être arrêté définitivement et une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit être instaurée.
En cas d'élévation de grade 2 ou 3 de la créatinine sanguine, Opdualag doit être suspendu et une corticothérapie à la dose de 0,5 à 1 mg/kg/jour de méthylprednisolone ou équivalent doit être instaurée. Après amélioration, Opdualag peut être repris après réduction progressive des corticoïdes. En cas d'aggravation ou d'absence d'amélioration malgré l'instauration d'une corticothérapie, la dose de corticoïdes doit être augmentée à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent, et Opdualag doit être arrêté définitivement.
Endocrinopathies d'origine immunologique
Des endocrinopathies sévères, incluant hypothyroïdie, hyperthyroïdie, insuffisance surrénalienne (incluant l'insuffisance corticosurrénalienne secondaire), hypophysite (incluant l'hypopituitarisme) et diabète sucré, ont été observées avec le nivolumab associé au relatlimab. Des cas d'acidocétose diabétique ont été observés avec le nivolumab en monothérapie et sont susceptibles de survenir avec le nivolumab associé au relatlimab (voir rubrique Effets indésirables).
Les patients doivent être surveillés pour déceler l'apparition de signes cliniques et de symptômes d'endocrinopathie et d'hyperglycémie, ou des modifications de la fonction thyroïdienne (au début du traitement, à intervalles réguliers en cours de traitement, et si cliniquement indiqué). Les patients peuvent présenter de la fatigue, des céphalées, des modifications de l'état mental, des douleurs abdominales, un transit intestinal inhabituel et une hypotension, ou des symptômes non spécifiques qui peuvent ressembler à d'autres causes telles que des métastases cérébrales ou une maladie sous-jacente. À moins qu'une autre étiologie n'ait été identifiée, les signes et symptômes d'endocrinopathie doivent être considérés comme d'origine immunologique.
Troubles de la thyroïde
En cas d'hypothyroïdie symptomatique, Opdualag doit être suspendu et une hormonothérapie thyroïdienne substitutive doit être instaurée, si nécessaire. En cas d'hyperthyroïdie symptomatique, Opdualag doit être suspendu et un traitement par antithyroïdiens doit être instauré, si nécessaire. Une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit également être envisagée en cas de suspicion d'une inflammation aiguë de la thyroïde. Après amélioration, Opdualag peut être repris après réduction progressive des corticoïdes, si nécessaire. La surveillance de la fonction thyroïdienne doit être poursuivie afin de s'assurer que le traitement hormonal substitutif approprié est utilisé. Opdualag doit être arrêté définitivement en cas d'hyperthyroïdie ou d'hypothyroïdie pouvant menacer le pronostic vital (grade 4).
Insuffisance surrénalienne
Opdualag doit être arrêté définitivement en cas d'insuffisance surrénalienne sévère (grade 3) ou pouvant menacer le pronostic vital (grade 4). En cas d'insuffisance surrénalienne symptomatique de grade 2, Opdualag doit être suspendu et une corticothérapie substitutive à une dose physiologique doit être instaurée, si nécessaire. La surveillance de la fonction surrénalienne et des taux d'hormones doit être poursuivie afin de s'assurer que la corticothérapie substitutive appropriée est utilisée.
Hypophysite
Opdualag doit être arrêté définitivement en cas d'hypophysite pouvant menacer le pronostic vital (grade 4). En cas d'hypophysite symptomatique de grade 2 ou 3, Opdualag doit être suspendu et un traitement hormonal substitutif doit être instauré, si nécessaire. En cas de suspicion d'inflammation aiguë de l'hypophyse, une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit également être envisagée. Après amélioration, Opdualag peut être repris après réduction progressive des corticoïdes, si nécessaire. La surveillance de la fonction hypophysaire et des taux d'hormones doit être poursuivie afin de s'assurer que le traitement hormonal substitutif approprié est utilisé.
Diabète sucré
En cas de diabète symptomatique, Opdualag doit être suspendu et un traitement substitutif par insuline doit être instauré, si nécessaire. La surveillance de la glycémie doit être poursuivie afin d'assurer que le traitement substitutif par insuline approprié est utilisé. Opdualag doit être arrêté définitivement en cas de diabète pouvant menacer le pronostic vital.
Effets indésirables cutanés d'origine immunologique
Des éruptions cutanées sévères ont été observées avec le nivolumab associé au relatlimab (voir rubrique Effets indésirables). Opdualag doit être suspendu en cas d'éruption cutanée de grade 3, et il doit être arrêté en cas d'éruption cutanée de grade 4. Les éruptions cutanées sévères doivent être prises en charge avec de hautes doses de corticoïdes, de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent.
De rares cas de SJS et de NET, dont certains d'issue fatale, ont été observés avec le nivolumab en monothérapie et sont susceptibles de survenir avec le nivolumab associé au relatlimab. En cas de suspicion de signes ou symptômes de SJS ou de NET, Opdualag doit être suspendu et le patient adressé à un service spécialisé pour évaluation et traitement. Si le patient reçoit un diagnostic confirmé de SJS ou de NET lors de l'utilisation d'Opdualag, l'arrêt définitif du traitement est recommandé (voir rubrique Posologie et mode d'administration).
L'utilisation d'Opdualag doit être considérée avec précaution chez un patient ayant présenté un effet indésirable cutané sévère ou ayant menacé le pronostic vital lors d'un précédent traitement anticancéreux stimulant l'immunité.
Myocardite d'origine immunologique
Des myocardites d'origine immunologique sévères ont été observées avec le nivolumab associé au relatlimab. Le diagnostic de myocardite exige un haut degré de suspicion. Les patients présentant des symptômes cardiaques ou cardio-pulmonaires doivent être évalués pour déterminer l'existence potentielle d'une myocardite. Si une myocardite est suspectée, l'administration immédiate d'une dose élevée de stéroïdes (prednisone 1 à 2 mg/kg/jour ou méthylprednisolone 1 à 2 mg/kg/jour) et une consultation immédiate en cardiologie avec un bilan diagnostique, selon les directives cliniques actuelles, doivent être initiées. Une fois qu'un diagnostic de myocardite est établi, Opdualag doit être suspendu ou arrêté définitivement, conformément aux indications ci-dessous.
En cas de myocardite de grade 3 ou 4, Opdualag doit être arrêté définitivement et une corticothérapie à la dose de 2 à 4 mg/kg/jour de méthylprednisolone ou équivalent doit être instaurée (voir rubrique Posologie et mode d'administration).
En cas de myocardite de grade 2, Opdualag doit être suspendu et une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit être instaurée. Après amélioration, la reprise d'Opdualag peut être envisagée après réduction progressive des corticoïdes. En cas d'aggravation ou d'absence d'amélioration malgré l'instauration d'une corticothérapie, les doses de corticoïdes doivent être augmentées à la dose de 2 à 4 mg/kg/jour de méthylprednisolone ou équivalent, et Opdualag doit être arrêté définitivement (voir rubrique Posologie et mode d'administration).
Autres effets indésirables d'origine immunologique
Les effets indésirables d'origine immunologique cliniquement significatifs suivants ont été rarement rapportés chez les patients traités par nivolumab en association avec le relatlimab : uvéite, pancréatite, syndrome de Guillain-Barré, myosite/rhabdomyolyse, encéphalite, anémie hémolytique et maladie de Vogt-Koyanagi-Harada (VKH).
Les effets indésirables d'origine immunologique cliniquement significatifs supplémentaires suivants ont été rarement rapportés avec le nivolumab en monothérapie ou avec le nivolumab associé à d'autres médicaments approuvés : démyélinisation, neuropathie auto-immune (incluant une parésie des nerfs facial et abducens), myasthénie grave, syndrome myasthénique, méningite aseptique, gastrite, sarcoïdose, duodénite, hypoparathyroïdie et cystite non infectieuse.
En cas de suspicion d'effets indésirables d'origine immunologique, une évaluation appropriée doit être effectuée afin de confirmer l'étiologie ou d'exclure d'autres causes. En fonction de la sévérité de l'effet indésirable, Opdualag doit être suspendu et des corticoïdes doivent être administrés. Après amélioration, Opdualag peut être repris après réduction progressive des corticoïdes. Opdualag doit être arrêté définitivement en cas d'effet indésirable sévère récurrent d'origine immunologique, et pour tout effet indésirable d'origine immunologique pouvant menacer le pronostic vital.
Autres mises en garde et précautions importantes, y compris effets de classe
Un rejet de greffe d'organe solide a été signalé après la mise sur le marché chez des patients traités par inhibiteurs de PD-1. Le traitement par nivolumab en association avec le relatlimab peut augmenter le risque de rejet chez les bénéficiaires d'une transplantation d'organe solide. Il convient de prendre en considération le rapport entre les bénéfices du traitement par nivolumab en association avec le relatlimab et le risque de rejet d'organe chez ces patients.
Des cas de lymphohistiocytose hémophagocytaire (LHH) ont été observés avec le nivolumab en monothérapie, avec le nivolumab associé au relatlimab ou avec le nivolumab associé à d'autres médicaments, dont un cas d'issue fatale avec le nivolumab associé au relatlimab. L'administration du nivolumab en association avec le relatlimab doit être effectuée avec précaution. Si une LHH est confirmée, l'administration du nivolumab en association avec le relatlimab doit être arrêtée et un traitement contre la LHH doit être instauré.
Chez les patients traités par nivolumab avant ou après une greffe allogénique de cellules souches hématopoïétiques (GCSH), des cas de maladie du greffon contre l'hôte (GVHD) sévères et d'apparition rapide, dont certains d'issue fatale, ont été rapportés. Le traitement par nivolumab en association avec le relatlimab peut augmenter le risque de GVHD sévère et de décès chez les patients ayant eu une GCSH allogénique antérieure, principalement chez ceux ayant un antécédent de GVHD. Il convient de prendre en considération le rapport entre les bénéfices du traitement par nivolumab en association avec le relatlimab et le risque de GVHD chez ces patients.
Réactions liées à la perfusion
Des réactions sévères liées à la perfusion ont été rapportées dans les études cliniques portant sur le nivolumab en association avec le relatlimab (voir rubrique Effets indésirables). En cas de réaction liée à la perfusion sévère ou pouvant menacer le pronostic vital, la perfusion d'Opdualag doit être arrêtée et un traitement médical approprié doit être administré. Les patients présentant une réaction liée à la perfusion d'intensité légère à modérée peuvent recevoir Opdualag sous surveillance étroite et un traitement préventif suivant les recommandations locales pour la prophylaxie des réactions liées à la perfusion.
Patients exclus de l'étude clinique pivot sur le mélanome avancé
Les patients présentant une maladie auto-immune active, un contexte médical nécessitant un traitement systémique impliquant la prise de doses modérées à élevées de corticoïdes ou de médicaments immunosuppresseurs, un mélanome uvéal, des métastases cérébrales ou leptoméningées actives ou non traitées, ou encore ceux présentant des antécédents de myocardite, une augmentation des taux de troponine supérieure à 2 fois la LSN ou un indice de performance de l'ECOG ≥ 2, ont été exclus de l'étude clinique pivot portant sur le nivolumab en association avec le relatlimab. En l'absence de données, le nivolumab en association avec le relatlimab doit être utilisé avec précaution dans ces populations, après évaluation attentive du rapport bénéfice/risque au cas par cas.
Carte patient
Le prescripteur doit discuter avec le patient des risques liés au traitement par Opdualag. Le patient recevra la carte patient et devra toujours la garder sur lui.
Résumé du profil de sécurité
Le nivolumab en association avec le relatlimab est associé à des effets indésirables d'origine immunologique (voir rubrique « Description des effets indésirables sélectionnés » ci-dessous). Les recommandations de prise en charge de ces effets indésirables sont décrites à la rubrique Mises en garde spéciales et précautions d'emploi.
Les effets indésirables les plus fréquents sont les suivants : fatigue (41 %), douleurs musculo-squelettiques (32 %), éruption cutanée (29 %), arthralgie (26 %), diarrhée (26 %), prurit (26 %), céphalées (20 %), nausées (19 %), toux (16 %), appétit diminué (16 %), hypothyroïdie (16 %), douleurs abdominales (14 %), vitiligo (13 %), fièvre (12 %), constipation (11 %), infection des voies urinaires (11 %), dyspnée (10 %) et vomissements (10 %).
Les effets indésirables graves les plus fréquents sont les suivants : insuffisance surrénalienne (1,4 %), anémie (1,4 %), dorsalgie (1,1 %), colite (1,1 %), diarrhée (1,1 %), myocardite (1,1 %), pneumonie (1,1 %) et infection des voies urinaires (1,1 %). L'incidence des effets indésirables de grade 3 à 5 chez les patients atteints d'un mélanome avancé (non résécable ou métastatique) était de 43 % pour le nivolumab en association avec le relatlimab et de 35 % chez les patients traités par nivolumab seul.
Liste tabulée des effets indésirables
La sécurité du nivolumab en association avec le relatlimab a été évaluée chez 355 patients atteints d'un mélanome avancé (non résécable ou métastatique) (étude CA224047). Les effets indésirables rapportés dans l'ensemble des données chez les patients traités par nivolumab en association avec le relatlimab, avec un suivi médian de 19,94 mois, sont présentés dans le Tableau 2. Les fréquences indiquées dans le Tableau 2 ci-dessus reposent sur les fréquences des effets indésirables toutes causes confondues. Ces effets indésirables sont présentés par classe de systèmes d'organes et par ordre de fréquence. Les fréquences sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre de gravité décroissant.
Tableau 2 : Effets indésirables observés dans le cadre des études cliniques
|
Infections et infestations |
|
|
Très fréquent |
infection des voies urinaires |
|
Fréquent |
infection des voies aériennes supérieures |
|
Peu fréquent |
folliculite |
|
Affections hématologiques et du système lymphatique |
|
|
Très fréquent |
anémiea, lymphopéniea, neutropéniea, leucopéniea |
|
Fréquent |
thrombopéniea, éosinophilie |
|
Peu fréquent |
anémie hémolytique |
|
Affections endocriniennes |
|
|
Très fréquent |
hypothyroïdie |
|
Fréquent |
insuffisance surrénalienne, hypophysite, hyperthyroïdie, thyroïdite |
|
Peu fréquent |
hypopituitarisme, hypogonadisme |
|
Troubles du métabolisme et de la nutrition |
|
|
Très fréquent |
appétit diminué |
|
Fréquent |
diabète sucré, hypoglycémiea, perte de poids, hyperuricémie, hypoalbuminémie, déshydratation |
|
Affections psychiatriques |
|
|
Fréquent |
état confusionnel |
|
Affections du système nerveux |
|
|
Très fréquent |
céphalées |
|
Fréquent |
neuropathie périphérique, sensations vertigineuses, dysgueusie |
|
Peu fréquent |
encéphalite, syndrome de Guillain-Barré, névrite optique |
|
Affections oculaires |
|
|
Fréquent |
uvéite, défauts visuels, sécheresse oculaire, augmentation de la sécrétion lacrymale |
|
Peu fréquent |
maladie de Vogt-Koyanagi-Harada, hyperhémie oculaire |
|
Affections cardiaques |
|
|
Fréquent |
myocardite |
|
Peu fréquent |
épanchement péricardique |
|
Affections vasculaires |
|
|
Fréquent |
phlébite |
|
Affections respiratoires, thoraciques et médiastinales |
|
|
Très fréquent |
dyspnée, toux |
|
Fréquent |
pneumopathie inflammatoireb, congestion nasale |
|
Peu fréquent |
asthme, épanchement pleural |
|
Affections gastro-intestinales |
|
|
Très fréquent |
diarrhée, vomissements, nausées, douleurs abdominales, constipation |
|
Fréquent |
colite, pancréatite, gastrite, dysphagie, stomatite, sécheresse buccale |
|
Peu fréquent |
œsophagite |
|
Rare |
insuffisance pancréatique exocrine |
|
Fréquence indéterminée |
maladie cœliaque |
|
Affections hépatobiliaires |
|
|
Fréquent |
hépatite |
|
Peu fréquent |
cholangite |
|
Affections de la peau et du tissu sous-cutané |
|
|
Très fréquent |
éruption cutanée, vitiligo, prurit |
|
Fréquent |
alopécie, kératose lichénoïde, réaction de photosensibilité, sécheresse cutanée |
|
Peu fréquent |
pemphigoïde, psoriasis, urticaire |
|
Affections musculo-squelettiques et systémiques |
|
|
Très fréquent |
douleurs musculo-squelettiques, arthralgie |
|
Fréquent |
arthrite, spasmes musculaires, faiblesse musculaire |
|
Peu fréquent |
myosite, syndrome de Sjögren, pseudopolyarthrite rhizomélique, polyarthrite rhumatoïde, lupus érythémateux systémique |
|
Affections du rein et des voies urinaires |
|
|
Fréquent |
insuffisance rénale, protéinurie |
|
Peu fréquent |
néphrite |
|
Affections des organes de reproduction et du sein |
|
|
Peu fréquent |
azoospermie |
|
Troubles généraux et anomalies au site d'administration |
|
|
Très fréquent |
fatigue, fièvre |
|
Fréquent |
œdème, syndrome grippal, frissons |
|
Rare |
sérite |
|
Investigations |
|
|
Très fréquent |
augmentation du taux d'ASATa, augmentation du taux d'ALATa, hyponatrémiea, augmentation du taux de créatininea, augmentation du taux de phosphatase alcalinea, hyperkaliémiea, hypocalcémiea, hypomagnésémiea, hypercalcémiea, hypokaliémiea |
|
Fréquent |
augmentation du taux de bilirubinea, hypernatrémiea, hypermagnésémiea, augmentation du taux de troponine, augmentation du taux de gamma-glutamyl transférase, augmentation du taux de lactate déshydrogénase sanguine, augmentation du taux de lipase, augmentation du taux d'amylase |
|
Peu fréquent |
augmentation du taux de protéine C-réactive, augmentation de la vitesse de sédimentation des érythrocytes |
|
Lésions, intoxications et complications liées aux procédures |
|
|
Fréquent |
réaction liée à la perfusion |
a Les fréquences représentent la proportion de patients ayant présenté une aggravation des valeurs biologiques par rapport aux valeurs à l'inclusion.
b Un cas d'issue fatale a été rapporté dans l'étude clinique.
Description des effets indésirables sélectionnés
Pneumopathie inflammatoire d'origine immunologique
Chez les patients traités par nivolumab en association avec le relatlimab, l’incidence des pneumopathies inflammatoires, incluant la pneumopathie interstitielle diffuse et l’infiltration pulmonaire, était de 5,1 %. L’incidence des événements de grade 3/4 était de 0,8 %. L’incidence des événements d’issue fatale était de 0,28 %. Le délai médian de survenue était de 28 semaines (de 3,6 à 94,4 semaines). Une résolution est survenue chez 83,3 % des patients avec un délai médian de résolution de 12,0 semaines (de 2,1 à 29,7+ semaines). La survenue d’une pneumopathie inflammatoire d’origine immunologique a entraîné l’arrêt définitif du nivolumab en association avec le relatlimab chez 1,7 % des patients et l’instauration d’une corticothérapie à forte dose (≥ 40 mg de prednisone ou équivalent par jour) chez 55,6 % des patients présentant une pneumopathie inflammatoire d’origine immunologique.
Colite d'origine immunologique
Chez les patients traités par nivolumab en association avec le relatlimab, 15,8 % ont présenté des diarrhées, des colites ou des selles fréquentes. L'incidence des événements de grade 3/4 était de 2,0 %. Le délai médian de survenue était de 14 semaines (de 0,1 à 95,6 semaines). Une résolution est survenue chez 92,7 % des patients avec un délai médian de résolution de 3,9 semaines (de 0,1 à 136,9+ semaines). La survenue d'une colite d'origine immunologique a entraîné l'arrêt définitif du nivolumab en association avec le relatlimab chez 2,0 % des patients et l'instauration d'une corticothérapie à forte dose (≥ 40 mg de prednisone ou équivalent par jour) chez 33,9 % des patients présentant une colite d'origine immunologique.
Hépatite d'origine immunologique
Chez les patients traités par nivolumab en association avec le relatlimab, 13,2 % ont présenté des tests hépatiques anormaux. L'incidence des événements de grade 3/4 était de 3,9 %. Le délai médian de survenue était de 11 semaines (de 2,0 à 144,9 semaines). Une résolution est survenue chez 78,7 % des patients avec un délai médian de résolution de 6,1 semaines (de 1,0 à 88,1+ semaines). La survenue d'une hépatite d'origine immunologique a entraîné l'arrêt définitif du nivolumab en association avec le relatlimab chez 2,0 % des patients et l'instauration d'une corticothérapie à forte dose chez 38,3 % des patients présentant une hépatite d'origine immunologique.
Néphrite et dysfonction rénale d'origine immunologique
Chez les patients traités par nivolumab en association avec le relatlimab, 4,5 % ont présenté une néphrite ou une dysfonction rénale. L'incidence des événements de grade 3/4 était de 1,4 %. Le délai médian de survenue était de 21 semaines (de 1,9 à 127,9 semaines). Une résolution est survenue chez 81,3 % des patients avec un délai médian de résolution de 8,1 semaines (de 0,9 à 91,6+ semaines). La survenue d'une néphrite et d'une dysfonction rénale d'origine immunologique a entraîné l'arrêt définitif du nivolumab en association avec le relatlimab chez 1,1 % des patients et l'instauration d'une corticothérapie à forte dose (≥ 40 mg de prednisone ou équivalent par jour) chez 25,0 % des patients présentant une néphrite et une dysfonction rénale d'origine immunologique.
Endocrinopathies d'origine immunologique
Chez les patients traités par nivolumab en association avec
le relatlimab, 26 % ont présenté une endocrinopathie.
Des troubles thyroïdiens, incluant l'hypothyroïdie et
l'hyperthyroïdie, sont survenus chez 20,8 % des patients. Aucun trouble thyroïdien
de grade 3/4 n'est survenu. Des insuffisances surrénaliennes (incluant des
insuffisances corticosurrénaliennes aiguës) sont survenues chez 4,8 % des
patients. Des insuffisances surrénaliennes de grade 3/4 sont survenues chez 1,4
% des patients. Aucun cas d'hypopituitarisme de grade 3/4 n'est survenu. Des
hypophysites sont survenues chez 1,1 % des patients. L'incidence des
hypophysites de grade 3/4 était de 0,3 %. Un diabète sucré (incluant le diabète
sucré de type 1) est survenu chez 0,3 % des patients. L'incidence des cas de
diabète sucré de grade 3/4 était de 0,3 %.
Le délai médian de survenue de ces endocrinopathies était de
13 semaines (de 1,0 à 73,0 semaines). Une résolution est survenue chez 27,7 %
des patients. Le délai de résolution allait de 0,4 à 176,0+ semaines. La survenue
d'endocrinopathies d'origine immunologique a entraîné l'arrêt définitif du
nivolumab en association avec le relatlimab chez 1,1 % des patients et
l'instauration d'une corticothérapie à forte dose (≥ 40 mg de prednisone ou
équivalent par jour) chez 7,4 % des patients présentant des endocrinopathies
d'origine immunologique.
Effets indésirables cutanés d'origine immunologique
Chez les patients traités par nivolumab en association avec le relatlimab, 45,1 % ont présenté une éruption cutanée, incluant prurit et vitiligo. L'incidence des événements de grade 3/4 était de 1,4 %. Le délai médian de survenue était de 8 semaines (de 0,1 à 116,4 semaines). Une résolution est survenue chez 47,5 % des patients. Le délai de résolution allait de 0,1 à 166,9+ semaines. La survenue d'effets indésirables cutanés d'origine immunologique a entraîné l'arrêt définitif du nivolumab en association avec le relatlimab chez 0,3 % des patients et l'instauration d'une corticothérapie à forte dose (≥ 40 mg de prednisone ou équivalent par jour) chez 3,8 % des patients présentant des effets indésirables cutanés d'origine immunologique.
Myocardite d'origine immunologique
Chez les patients traités par nivolumab en association avec le relatlimab, 1,4 % ont présenté une myocardite. L’incidence des événements de grade 3/4 était de 0,6 %. Le délai médian de survenue était de 4,14 semaines (de 2,1 à 6,3 semaines). Une résolution est survenue chez 100 % des patients, avec un délai médian de résolution de 3 semaines (de 1,9 à 14,0 semaines). La survenue d’une myocardite a entraîné l’arrêt définitif du nivolumab en association avec le relatlimab chez 1,4 % des patients et l’instauration d’une corticothérapie à forte dose (≥ 40 mg de prednisone ou équivalent par jour) chez 100 % des patients présentant une myocardite d’origine immunologique.
Réactions liées à la perfusion
Chez les patients traités par nivolumab en association avec le relatlimab, 6,8 % ont présenté des réactions d'hypersensibilité/liées à la perfusion. Il s'agissait exclusivement d'événements de grade 1/2.
Tests de laboratoire anormaux
Chez les patients traités par nivolumab en association avec le relatlimab, la proportion de patients ayant présenté une modification des valeurs biologiques par rapport aux valeurs à l'inclusion vers une anomalie de grade 3 ou 4 a été la suivante : 3,6 % pour les anémies, 5,2 % pour les lymphopénies, 0,3 % pour les neutropénies, 0,6 % pour les augmentations du taux de phosphatase alcaline, 2,9 % pour les augmentations du taux d'ASAT, 3,5 % pour les augmentations du taux d'ALAT, 0,3 % pour les augmentations du taux de bilirubine totale, 0,9 % pour les augmentations du taux de créatinine, 1,5 % pour les hyponatrémies, 1,8 % pour les hyperkaliémies, 0,3 % pour les hypokaliémies, 0,9 % pour les hypercalcémies, 0,6 % pour les hypocalcémies, 0,9 % pour les hypermagnésémies et 0,6 % pour les hypomagnésémies.
Immunogénicité
Dans l'étude CA224047, parmi les patients évaluables pour les anticorps anti-médicaments, l'incidence des anticorps anti-relatlimab apparus pendant le traitement et des anticorps neutralisants dirigés contre le relatlimab dans le groupe traité par Opdualag était respectivement de 5,6 % (17/301) et de 0,3 % (1/301). L'incidence des anticorps anti-nivolumab apparus pendant le traitement et des anticorps neutralisants dirigés contre le nivolumab dans le groupe traité par Opdualag était respectivement de 4,0 % (12/299) et de 0,3 % (1/299), c'est-à-dire semblable à celle du groupe traité par nivolumab, soit 6,7 % (19/283) et 0,4 % (1/283), respectivement. Aucune altération du profil pharmacocinétique, d'efficacité ou de sécurité n'a été mise en évidence du fait de la formation d'anticorps anti-nivolumab ou anti-relatlimab.
Populations particulières
Patients âgés
Aucune différence globale en matière de sécurité n'a été rapportée entre les patients âgés (≥ 65 ans) et les patients plus jeunes (voir rubrique Propriétés pharmacodynamiques).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
Analyse de l'expression de
PD-L1 : la sélection des patients pouvant ce traitement repose sur le
niveau d'expression tumorale de PD-L1, confirmée par un test validé.
SURVEILLANCE continue de l'apparition
d'effets indésirables d'origine immunologique pendant et
Jusqu'à au moins 5 mois après
la dernière perfusion.
Femme en âge de PROCRER :
utiliser une contraception efficace pendant le traitement et jusqu' à 5 mois
après la dernière perfusion.
Femmes en âge de procréer/Contraception
L'utilisation d'Opdualag n'est pas recommandée chez les femmes en âge de procréer n'utilisant pas une contraception efficace, à moins que le bénéfice clinique attendu ne dépasse le risque potentiel. Une contraception efficace doit être poursuivie pendant au moins 5 mois après la dernière dose d'Opdualag.
Grossesse
Il existe des données limitées sur l'utilisation du nivolumab en association avec le relatlimab chez la femme enceinte. D'après son mécanisme d'action et les données issues des études effectuées chez l'animal, le nivolumab en association avec le relatlimab peut nuire au développement du fœtus lorsqu'il est administré chez la femme enceinte. Les études effectuées chez des animaux traités par nivolumab ont mis en évidence une toxicité embryofœtale (voir rubrique Données de sécurité préclinique). L'IgG4 humaine est connue pour traverser la barrière placentaire et le nivolumab et le relatlimab sont des IgG4 ; par conséquent, il existe un risque potentiel de transmission du nivolumab ou du relatlimab de la mère vers le fœtus. L'utilisation d'Opdualag n'est pas recommandée chez la femme enceinte ou en âge de procréer n'utilisant pas de contraception efficace, à moins que le bénéfice clinique attendu ne dépasse le risque potentiel.
Allaitement
On ne sait pas si le nivolumab et/ou le relatlimab sont excrétés dans le lait maternel. Les IgG humaines sont excrétées dans le lait maternel au cours des premiers jours suivant la naissance, mais leur concentration diminue rapidement jusqu'à un niveau faible. Ainsi, un risque pour le nourrisson allaité ne peut être exclu pendant cette courte période. Par la suite, Opdualag peut être utilisé pendant l'allaitement si nécessaire.
Fertilité
Aucune étude permettant d'évaluer l'effet du nivolumab et/ou du relatlimab sur la fertilité n'a été effectuée. En conséquence, l'effet du nivolumab et/ou du relatlimab sur la fertilité masculine et féminine n'est pas connu.
Le nivolumab et le relatlimab sont tous deux des anticorps monoclonaux humains. Ainsi, aucune étude n'a été réalisée sur les interactions. Les anticorps monoclonaux humains n'étant pas métabolisés par les enzymes du cytochrome P450 (CYP) ou d'autres enzymes métabolisant les substances actives, l'inhibition ou l'induction de ces enzymes lors de la co-administration de médicaments ne devraient pas entraîner de modification des paramètres pharmacocinétiques du relatlimab ou du nivolumab.
Compte tenu de l'absence de modulation significative des cytokines par le nivolumab et le relatlimab et donc, de l'absence d'influence sur l'expression des enzymes du cytochrome P450, le nivolumab et le relatlimab ne devraient pas entraîner de modification des paramètres pharmacocinétiques des autres substances actives métabolisées par les enzymes CYP.
Immunosuppression systémique
L'utilisation de corticoïdes systémiques et d'autres immunosuppresseurs à l'inclusion doit être évitée avant de commencer le nivolumab en association avec le relatlimab, du fait de leur interférence potentielle avec l'activité pharmacodynamique. Cependant, les corticoïdes systémiques et d'autres immunosuppresseurs peuvent être utilisés après l'instauration du nivolumab en association avec le relatlimab pour traiter les effets indésirables d'origine immunologique.
Le traitement doit être instauré et surveillé par un médecin expérimenté dans le traitement du cancer.
Les patients traités par Opdualag doivent recevoir la carte patient et être informés des risques liés à l'utilisation d'Opdualag (voir également la notice).
Analyse de l'expression de PD-L1
La sélection des patients pouvant recevoir le traitement par Opdualag doit reposer sur le niveau d'expression tumorale de PD-L1, confirmée par un test validé (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacodynamiques).
Posologie
La dose recommandée pour les adultes et les adolescents âgés de 12 ans et plus est de 480 mg de nivolumab et 160 mg de relatlimab toutes les 4 semaines, administré en perfusion intraveineuse pendant 30 minutes. Cette dose est établie pour les patients adolescents pesant au moins 30 kg (voir rubrique Propriétés pharmacocinétiques).
Le traitement par Opdualag doit être poursuivi tant qu'un bénéfice clinique est observé, ou jusqu'à ce que le patient ne tolère plus le traitement. Les augmentations ou diminutions de doses ne sont pas recommandées. Des administrations différées ou des interruptions de traitement peuvent être nécessaires selon la sécurité et la tolérance de chaque patient. Les recommandations concernant l'arrêt définitif du traitement ou la suspension des doses sont décrites dans le Tableau 1. Des recommandations détaillées pour la prise en charge des effets indésirables d'origine immunologique sont présentées à la rubrique Mises en garde spéciales et précautions d'emploi.
Tableau 1 : Recommandations de modification du traitement par Opdualag
|
Effet indésirable d'origine immunologique |
Sévérité |
Modification de traitement |
|
Pneumopathie inflammatoire d'origine immunologique |
Pneumopathie de grade 2 |
Suspendre la ou les doses jusqu'à la résolution des symptômes, l'amélioration des anomalies radiographiques, et la fin du traitement par corticoïdes |
|
Pneumopathie de grade 3 ou 4 |
Arrêter définitivement le traitement |
|
|
Colite d'origine immunologique |
Diarrhée ou colite de grade 2 ou 3 |
Suspendre la ou les doses jusqu'à la résolution des symptômes et la fin du traitement par corticoïdes, s'il s'est avéré nécessaire |
|
Diarrhée ou colite de grade 4 |
Arrêter définitivement le traitement |
|
|
Hépatite d'origine immunologique |
Augmentation des aspartate aminotransférases (ASAT) ou des alanine aminotransférases (ALAT) supérieure à 3 fois et jusqu'à 5 fois la limite supérieure de la normale (LSN) ou Augmentation de la bilirubine totale supérieure à 1,5 fois et jusqu'à 3 fois la LSN |
Suspendre la ou les doses jusqu'au retour des valeurs biologiques aux valeurs initiales et jusqu'à la fin du traitement par corticoïdes, s'il s'est avéré nécessaire |
|
Augmentation des ASAT ou ALAT supérieure à 5 fois la LSN, indépendamment de la valeur de référence ou Augmentation de la bilirubine totale supérieure à 3 fois la LSN ou Augmentation concomitante des ASAT ou ALAT supérieure à 3 fois la LSN et augmentation de la bilirubine totale supérieure à 2 fois la LSN |
Arrêter définitivement le traitement |
|
|
Néphrite et dysfonction rénale d'origine immunologique |
Élévation de la créatinine de grade 2 ou 3 |
Suspendre la ou les doses jusqu'au retour de la créatinine à la valeur initiale et jusqu'à la fin du traitement par corticoïdes |
|
Élévation de la créatinine de grade 4 |
Arrêter définitivement le traitement |
|
|
Endocrinopathies d'origine immunologique |
Hypothyroïdie, hyperthyroïdie, hypophysite symptomatiques de grade 2 ou 3 Insuffisance surrénalienne de grade 2 Diabète de grade 3 |
Suspendre la ou les doses jusqu'à la résolution des symptômes et la fin du traitement par corticoïdes (s'il s'est avéré nécessaire pour les symptômes d'une inflammation aiguë). Le traitement doit être poursuivi en cas de traitement hormonal substitutifa tant qu'il n'y a pas de symptômes |
|
Hypothyroïdie de grade 4 Hyperthyroïdie de grade 4 Hypophysite de grade 4 Insuffisance surrénalienne de grade 3 ou 4 Diabète de grade 4 |
Arrêter définitivement le traitement |
|
|
Effets indésirables cutanés d'origine immunologique |
Éruption cutanée de grade 3 |
Suspendre la ou les doses jusqu'à la résolution des symptômes et la fin du traitement par corticoïdes |
|
Suspicion de syndrome de Stevens-Johnson (SJS) ou de nécrolyse épidermique toxique (NET) |
Suspendre la ou les doses |
|
|
Éruption cutanée de grade 4 Confirmation de SJS/NET |
Arrêter définitivement le traitement (voir rubrique Mises en garde spéciales et précautions d'emploi) |
|
|
Myocardite d'origine immunologique |
Myocardite de grade 2 |
Suspendre la ou les doses jusqu'à la résolution des symptômes et la fin du traitement par corticoïdesb |
|
Myocardite de grade 3 ou 4 |
Arrêter définitivement le traitement |
|
|
Autres effets indésirables d'origine immunologique |
Grade 3 (première apparition) |
Suspendre la ou les doses |
|
Grade 4 ou grade 3 récidivant ; grade 2 ou 3 persistant malgré une modification de traitement ; impossibilité de réduire la dose de corticoïdes à 10 mg de prednisone ou équivalent par jour |
Arrêter définitivement le traitement |
Remarque : les grades de toxicité correspondent à la classification du National Cancer Institute Common Terminology Criteria for Adverse Events Version 5.0 (NCI-CTCAE v5).
a La recommandation pour l'utilisation d'un traitement hormonal substitutif est fournie à la rubrique Mises en garde spéciales et précautions d'emploi.
b La tolérance de la reprise du traitement par Opdualag chez les patients ayant présenté précédemment une myocardite d'origine immunologique, n'est pas connue.
Populations particulières
Population pédiatrique
La sécurité et l'efficacité d'Opdualag chez les enfants âgés de moins de 12 ans n'ont pas été établies. Aucune donnée n'est disponible (voir rubrique Propriétés pharmacocinétiques).
Patients âgés
Aucune adaptation posologique n'est nécessaire chez les patients âgés (≥ 65 ans) (voir rubrique Propriétés pharmacocinétiques).
Insuffisance rénale
Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance rénale légère ou modérée (voir rubrique Propriétés pharmacocinétiques). Les données des patients présentant une insuffisance rénale sévère sont trop limitées pour tirer des conclusions dans cette population.
Insuffisance hépatique
Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère ou modérée (voir rubrique Propriétés pharmacocinétiques). Les données des patients présentant une insuffisance hépatique sévère sont trop limitées pour tirer des conclusions dans cette population.
Mode d'administration
Opdualag doit être exclusivement administré par voie intraveineuse. Il doit être administré en perfusion intraveineuse sur une période de 30 minutes.
Opdualag ne doit être administré ni en injection rapide ni en bolus intraveineux.
Opdualag peut être utilisé sans dilution ou être dilué dans une solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) ou une solution injectable de glucose à 50 mg/mL (5 %) (voir rubrique Précautions particulières d’élimination et de manipulation).
Pour les instructions concernant la préparation et la manipulation du médicament avant administration, voir la rubrique Précautions particulières d’élimination et de manipulation.
Durée de conservation :
Flacon non ouvert
3 ans.
Après préparation de la perfusion
La stabilité chimique et physique en cours d'utilisation, depuis le moment de la préparation, a été démontrée comme suit (les délais incluent la période d'administration) :
|
Préparation de la perfusion |
Stabilité chimique et physique en cours d'utilisation |
|
|
Conservation entre 2 °C et 8 °C à l'abri de la lumière |
Conservation à température ambiante (≤ 25 °C) et lumière ambiante |
|
|
Non diluée ou diluée dans une solution injectable de chlorure de sodium à glucose à 50 mg/mL (5 %) |
30 jours |
24 heures (sur un total de 30 jours de conservation) |
| Diluée dans une solution injectable de glucose à 50 mg/mL (5 %) |
7 jours |
24 heures (sur un total de 7 jours de conservation) |
D'un point de vue microbiologique, la solution pour perfusion préparée doit être utilisée immédiatement, indépendamment du diluant. Si elle n'est pas utilisée immédiatement, les durées et les conditions de conservation avant utilisation sont sous la responsabilité de l'utilisateur et ne doivent normalement pas dépasser 24 heures entre 2 °C et 8 °C, sauf si la préparation a eu lieu dans des conditions d'asepsie validées et contrôlées (voir rubrique Précautions particulières d’élimination et de manipulation).
Précautions particulières de conservation :
À conserver au réfrigérateur (entre 2 °C et 8 °C).
Ne pas congeler.
Conserver le flacon dans l'emballage extérieur, à l'abri de
la lumière.
Les flacons non ouverts peuvent être conservés à température
ambiante contrôlée (jusqu'à 25 °C) jusqu'à 72 heures.
Pour les conditions de conservation après préparation de la
perfusion, voir la rubrique Durée de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments. Opdualag ne doit pas être perfusé simultanément avec d'autres médicaments sur la même ligne intraveineuse.
En cas de surdosage, les patients doivent être étroitement surveillés à la recherche de signes ou symptômes évocateurs d'effets indésirables, et un traitement symptomatique approprié doit être instauré immédiatement.
Classe pharmacothérapeutique : agents antinéoplasiques, anticorps monoclonaux, Code ATC : L01FY02.
Mécanisme d'action
Opdualag est une association à dose fixe de nivolumab, un inhibiteur anti-PD-1 (« programmed death-1 »), et de relatlimab, un inhibiteur anti-LAG-3 (« lymphocyte-activation gene-3 »).
La liaison des ligands PD-1, PD-L1 et PD-L2 au récepteur PD-1 situé sur les lymphocytes T inhibe la prolifération des lymphocytes T et la production de cytokines. La régulation à la hausse des ligands PD-1 survient dans certaines tumeurs et la transmission du signal par cette voie peut contribuer à l'inhibition de la surveillance immunitaire des lymphocytes T actifs des tumeurs. Le nivolumab est un anticorps monoclonal humain de type IgG4 qui se lie au récepteur PD-1, bloque son interaction avec ses ligands PD-L1 et PD-L2, et limite l'inhibition médiée par la voie PD-1 de la réponse immunitaire, y compris de la réponse immunitaire anti-tumorale. Dans des modèles de tumeurs murines syngéniques, le blocage de l'activité de PD-1 a entraîné une diminution de la croissance de la tumeur.
Le relatlimab est un anticorps monoclonal humain de type IgG4 qui se lie au récepteur LAG-3, bloque son interaction avec ses ligands, y compris avec le MHC II, et limite l'inhibition médiée par la voie LAG-3 de la réponse immunitaire. L'antagonisme de cette voie favorise la prolifération des lymphocytes T et la libération de cytokines.
L'association du nivolumab (anti-PD-1) et du relatlimab (anti-LAG-3) entraîne une activation accrue des lymphocytes T par rapport à l'activité des deux anticorps seuls. Dans des modèles de tumeurs murines syngéniques, le blocage de LAG-3 potentialise l'activité anti-tumorale du blocage de PD-1, inhibant ainsi la croissance de la tumeur et favorisant sa régression.
Efficacité et sécurité cliniques
Étude de phase 2/3, randomisée, comparant le nivolumab en association avec le relatlimab versus le nivolumab seul chez des patients atteints de mélanome non résécable ou métastatique non précédemment traités (CA224047)
La sécurité et l'efficacité du nivolumab en association avec le relatlimab dans le traitement des patients atteints de mélanome non résécable ou métastatique non précédemment traité ont été évaluées dans une étude de phase 2/3, randomisée, en double aveugle (CA224047). L'étude a inclus des patients dont l'indice de performance de l'ECOG était de 0 ou 1, et atteints d'un mélanome confirmé histologiquement de stade III (non résécable) ou IV selon l'American Joint Committee on Cancer (AJCC), 8e édition. Les patients pouvaient avoir reçu un traitement antérieur adjuvant ou néoadjuvant pour le mélanome (un traitement anti-PD-1, anti-CTLA-4 ou BRAF-MEK était autorisé à condition qu'un délai minimum de 6 mois se soit écoulé entre la dernière dose du traitement et la date de la récidive ; un traitement par interféron était autorisé à condition qu'un délai minimum de 6 semaines se soit écoulé entre la dernière dose du traitement et la randomisation). Les patients présentant une maladie auto-immune active, des antécédents de myocardite, une augmentation des taux de troponine supérieure à 2 fois la LSN ou un indice de performance de l'ECOG ≥ 2, un contexte médical nécessitant un traitement systémique impliquant la prise de doses modérées à élevées de corticoïdes ou de médicaments immunosuppresseurs, un mélanome uvéal, des métastases cérébrales ou leptoméningées actives ou non traitées, ont été exclus de l'étude (voir rubrique Mises en garde spéciales et précautions d'emploi).
Un total de 714 patients ont été randomisés pour recevoir du nivolumab en association avec le relatlimab (n = 355) ou du nivolumab seul (n = 359). Les patients du bras traité par l'association ont reçu 480 mg de nivolumab/160 mg de relatlimab pendant 60 minutes, toutes les 4 semaines. Les patients du bras traité par nivolumab seul ont reçu 480 mg de nivolumab toutes les 4 semaines. La randomisation a été stratifiée par niveau d'expression tumorale de PD-L1 (≥ 1 % versus < 1) à l'aide du test PD-L1 IHC 28-8 pharmDx, par niveau d'expression de LAG-3 (≥ 1 % versus < 1), tel que déterminé par un dosage LAG-3 IHC analytiquement validé, par statut de la mutation BRAF V600 et par stade métastatique tel que défini dans le système de classification de l'AJCC, 8e édition (M0/M1a[0] versus M1a[1]). Les patients ont reçu le traitement jusqu'à progression de la maladie ou apparition d'une toxicité inacceptable. Des évaluations de la tumeur ont été effectuées selon les critères RECIST (Response Evaluation Criteria in Solid Tumours), version 1.1, 12 semaines après la randomisation puis toutes les 8 semaines durant les 52 premières semaines, et toutes les 12 semaines jusqu'à progression de la maladie ou arrêt du traitement, selon la dernière occurrence. Le critère d'évaluation principal de l'efficacité était la survie sans progression (SSP), déterminée par un comité de revue centralisé indépendant en aveugle (RCIA). Les critères d'évaluation secondaires de l'efficacité étaient la survie globale (SG) et le taux de réponse global (ORR), déterminés par RCIA. La hiérarchisation statistique des tests s'est établie comme suit : SSP suivie de la SG puis de l'ORR. Les critères d'évaluation principaux et secondaires ont été évalués dans la population en intention de traiter (ITT). Aucun test formel de l'ORR n'a été effectué étant donné qu'une comparaison formelle de la SG n'était pas statistiquement significative.
Les caractéristiques de la population ITT à l'inclusion étaient comparables entre les deux groupes. L'âge médian était de 63 ans (de 20 à 94 ans), dont 47 % ≥ 65 ans et 19 % ≥ 75 ans. La majorité des patients étaient de type caucasien (97 %) et de sexe masculin (58 %). L'indice de performance de l'ECOG à l'inclusion était de 0 (67 %) ou 1 (33 %). La majorité des patients étaient atteints d'une maladie de stade IV selon la classification de l'AJCC (92 %) ; 38,9 % étaient atteints d'une maladie classée M1c, 2,4 % étaient atteints d'une maladie classée M1d, 8,7 % avaient précédemment reçu des traitements systémiques et 36 % présentaient un taux de LDH de référence supérieur à la LSN lors de l'admission à l'étude. Trente-neuf pour cent des patients présentaient un mélanome avec une mutation BRAF positive, 75 % des patients présentaient une expression de LAG-3 ≥ 1 % et 41 % des patients présentaient une expression de PD-L1 ≥ 1 % sur la membrane de la cellule tumorale. Parmi les patients présentant une expression tumorale de PD-L1 quantifiable, la répartition entre les deux groupes de traitement était équilibrée. Chez les patients présentant un niveau d'expression de PD-L1 inférieur à 1 %, les données démographiques et les caractéristiques de la maladie à l'inclusion étaient généralement comparables entre les bras de traitement.
Lors de l'analyse
primaire de la population ITT, avec un suivi médian de 13,21 mois (de 0 à 33,1
mois), une amélioration statistiquement significative de la SSP a été observée,
avec une SSP médiane de 10,12 mois dans le groupe traité par nivolumab en
association avec le relatlimab, versus 4,63 mois dans le groupe traité
par nivolumab seul (HR = 0,75 ; IC à 95 % : 0,62 ; 0,92 ; p = 0,0055).
Lors de l'analyse finale prédéfinie de la SG dans la population ITT, avec un
suivi médian de 19,3 mois, la SG n'a pas été significative sur le plan
statistique (HR = 0,80 ; IC à 95 % : 0,64 ; 1,01).
Analyse de sous-groupes prédéfinis avec niveau d'expression de PD-L1 inférieur à 1 %
Les principaux résultats d'efficacité pour le sous-groupe de patients présentant un niveau d'expression tumorale de PD-L1 inférieur à 1 %, issus d'une analyse exploratoire avec un suivi médian de 17,78 mois (de 0,26 à 40,64 mois), sont résumés dans le Tableau 3.
Tableau 3 : Résultats d'efficacité chez les patients dont le niveau d'expression de PD-L1 par
les cellules tumorales est inférieur à 1 % (CA224047)

a Hazard ratio basé sur le modèle de risque proportionnel de Cox non stratifié.
b Les résultats en matière de SG ne sont pas encore matures.
Durée médiane de suivi : 17,78 mois.
NA = non atteint.
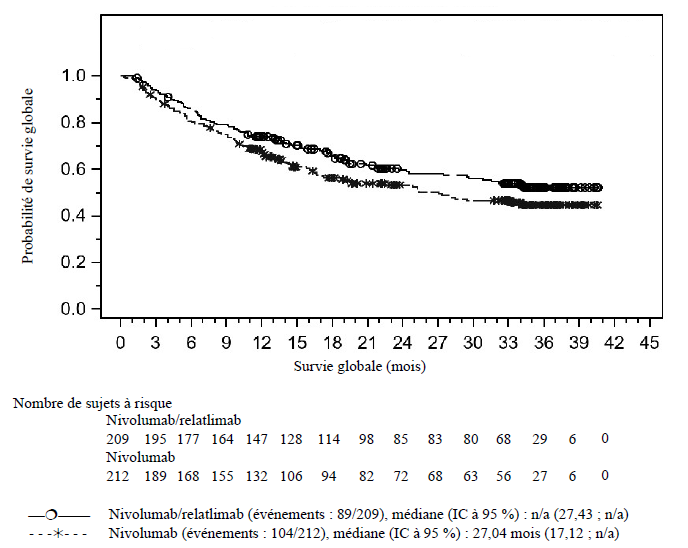
Les courbes de Kaplan-Meier illustrant la SSP et la SG chez les patients présentant un niveau d'expression de PD-L1 par les cellules tumorales inférieur à 1 % sont présentées respectivement dans les Figures 1 et 2.
Figure 1 : Courbes de Kaplan-Meier illustrant la SSP chez les patients présentant un niveau d'expression de PD-L1 par les cellules tumorales inférieur à 1 % (CA224047)
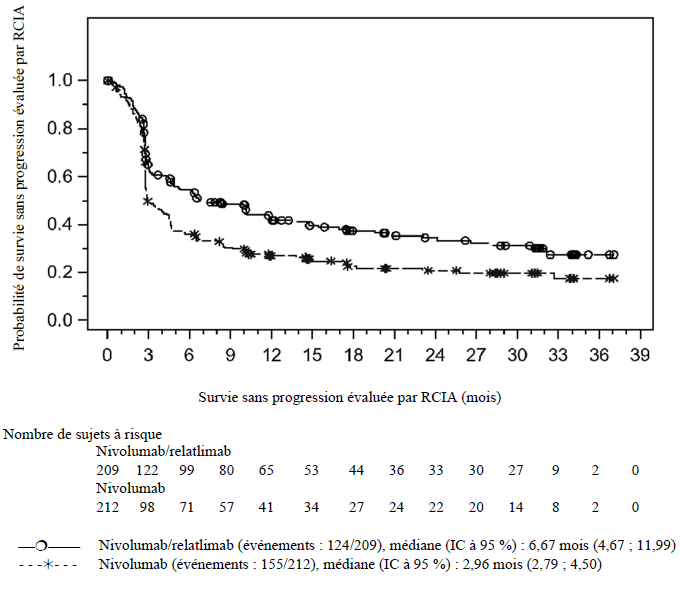
Figure 2 : Courbes de Kaplan-Meier illustrant la survie globale chez les patients présentant un niveau d'expression de PD-L1 par les cellules tumorales inférieur à 1 % (CA224047)
La pharmacocinétique du relatlimab à la suite de l'administration du nivolumab en association avec le relatlimab a été caractérisée chez des patients atteints de différents types de cancers ayant reçu des doses de relatlimab de 20 à 800 mg toutes les 2 semaines et de 160 à 1 440 mg toutes les 4 semaines, en monothérapie ou en association avec des doses de nivolumab de 80 ou 240 mg toutes les 2 semaines ou de 480 mg toutes les 4 semaines.
L'état d'équilibre des concentrations de relatlimab a été atteint sous 16 semaines avec un traitement toutes les 4 semaines, et l'accumulation systémique a été multipliée par 1,9. La concentration moyenne (Cmoy) de relatlimab après la première dose a donné lieu à une augmentation proportionnelle de la dose jusqu'à des doses ≥ 160 mg toutes les 4 semaines.
Tableau 4 : Moyenne géométrique (CV %) des expositions à l'état d'équilibre du nivolumab et du relatlimab suite à l'administration toutes les 4 semaines d'une dose fixe de 480 mg de nivolumab en association avec 160 mg de relatlimab
|
|
Cmax (μg/mL) |
Cmin (μg/mL) |
Cmoy (μg/mL) |
|
Relatlimab |
62,2 (30,1) |
15,3 (64,3) |
28,8 (44,8) |
|
Nivolumab |
187 (32,9) |
59,7 (58,6) |
94,4 (43,3) |
Sur la base des analyses pharmacocinétiques de population, il a été défini que des durées de perfusion de 30 et 60 minutes avec l'association à dose fixe de nivolumab et de relatlimab produiraient une exposition similaire (< 1 % de différence) au nivolumab et au relatlimab.
Dans l'étude CA224047, la moyenne géométrique Cmin du nivolumab à l'état d'équilibre dans le bras traité par nivolumab en association avec le relatlimab était similaire à celle du bras traité par nivolumab seul, avec un rapport de moyenne géométrique de 0,931 (IC à 95 % : 0,855 - 1,013).
Distribution
La valeur de la moyenne géométrique (CV %) pour le volume de distribution du nivolumab à l'état d'équilibre est de 6,65 L (19,2 %), et de 6,65 L (19,8 %) pour le relatlimab.
Biotransformation
Le nivolumab et le relatlimab sont des anticorps monoclonaux thérapeutiques de type IgG4 conçus pour être catabolisés en peptides de petite taille, en acides aminés et en glucides de petite taille par le lysosome ou l'endocytose médiée par le récepteur.
Élimination
La clairance du nivolumab à l'état d'équilibre est de 21,1 % inférieure (moyenne géométrique [CV %], 7,57 mL/h [40,1 %]) à la clairance enregistrée après la première dose (9,59 mL/h [40,3 %]) et la demi-vie terminale (t1/2) est de 26,5 jours (36,4 %).
La clairance du relatlimab à l'état d'équilibre est de 9,7 % inférieure (moyenne géométrique [CV %], 5,48 mL/h [41,3 %]) à la clairance enregistrée après la première dose (6,06 mL/h [38,9 %]). Suite à l'administration toutes les 4 semaines de 160 mg de relatlimab et de 480 mg de nivolumab, la moyenne géométrique (CV %) de la demi-vie effective (t1/2) du relatlimab est de 26,2 jours (37 %).
Populations particulières
Une analyse de pharmacocinétique de population suggère que les facteurs suivants n'ont pas d'effet cliniquement important sur la clairance du nivolumab et du relatlimab : âge (de 17 à 92 ans), sexe (masculin [1 056] et féminin [657]), origine ethnique (caucasienne [1 655], afro-américaine [167] et asiatique [41]). Le poids du patient (de 37 à 170 kg) était une covariable significative dans les analyses de pharmacocinétique du nivolumab et du relatlimab. Cependant, il n'existe aucun impact cliniquement pertinent lié à l'analyse de la réponse à l'exposition.
Population pédiatrique
Des données limitées suggèrent que la clairance et le volume de distribution du nivolumab chez les sujets adolescents atteints de tumeurs solides étaient respectivement de 36 % et 16 % inférieurs à ceux des patients adultes de référence. On ignore s'il en va de même pour les patients atteints de mélanome et si la clairance et le volume de distribution du relatlimab sont également plus bas chez les adolescents que chez les adultes. Toutefois, d'après des simulations de pharmacocinétique de population, l'exposition au nivolumab et au relatlimab chez les adolescents pesant au moins 30 kg devrait entraîner des résultats de sécurité et d'efficacité comparables à ceux observés chez les adultes de même poids, pour une dose recommandée identique.
Insuffisance rénale
L'effet de l'insuffisance rénale sur la clairance du nivolumab et du relatlimab a été évalué par le biais d'une analyse de pharmacocinétique de population chez des patients présentant une insuffisance rénale légère ou modérée, en comparaison avec des patients présentant une fonction rénale normale. Aucune différence cliniquement importante de la clairance du nivolumab ou du relatlimab n'a été observée entre les patients présentant une insuffisance rénale et les patients présentant une fonction rénale normale.
Insuffisance hépatique
L'effet de l'insuffisance hépatique sur la clairance du nivolumab et du relatlimab a été évalué par le biais d'une analyse de pharmacocinétique de population chez des patients présentant une insuffisance hépatique légère (bilirubine totale [BT] inférieure ou égale à la limite supérieure de la normale [LSN] et ASAT supérieures à la LSN ou BT supérieure à 1 à 1,5 fois la LSN, indépendamment des ASAT) ou modérée (BT supérieure à 1,5 à 3 fois la LSN, indépendamment des ASAT), en comparaison avec des patients présentant une fonction hépatique normale. Aucune différence cliniquement importante de la clairance du nivolumab ou du relatlimab n'a été observée entre les patients présentant une insuffisance hépatique et les patients présentant une fonction hépatique normale.
Immunogénicité
Le faible taux d'incidence observé en matière d'anticorps anti-nivolumab et anti-relatlimab apparus pendant le traitement n'a eu aucun effet sur la pharmacocinétique du nivolumab et du relatlimab.
Opdualag a une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines. Du fait des effets indésirables potentiels, tels que la fatigue et les sensations vertigineuses (voir rubrique Effets indésirables), les patients doivent être prévenus qu'ils doivent être prudents lorsqu'ils conduisent ou utilisent des machines, tant qu'ils ne sont pas certains qu'Opdualag n'altère pas leur vigilance.
Nivolumab en association avec le relatlimab
Aucune étude animale n'a été menée avec le nivolumab associé au relatlimab pour évaluer le risque de carcinogénicité, de génotoxicité et de toxicité pour la reproduction et pour le développement.
Dans le cadre d'une étude menée sur un mois chez des singes recevant du nivolumab et du relatlimab, une inflammation au niveau du système nerveux central (plexus choroïdes, système vasculaire, méninges, moelle épinière) et de l'appareil reproducteur (épididyme, vésicules séminales et testicules) a été observée. Bien qu'aucune marge de sécurité n'ait été établie concernant ces effets observés avec l'association, ceux-ci surviennent à des doses supposant des niveaux d'exposition nettement supérieurs (13 fois pour le nivolumab et 97 fois pour le relatlimab) à ceux atteints chez les patients.
Relatlimab
Aucune donnée n'est disponible chez l'animal concernant l'effet du relatlimab sur la grossesse et la reproduction. Dans le cadre d'une étude sur la toxicité embryofœtale menée chez des souris exposées à un anticorps murins anti-LAG3, aucun effet maternel ou sur le développement n'a été observé. Les effets du relatlimab sur le développement prénatal et postnatal n'ont pas été évalués. Toutefois, d'après son mécanisme d'action, le blocage de LAG-3 par le relatlimab peut avoir un effet négatif sur la grossesse similaire à celui du nivolumab. Aucune étude de fertilité n'a été effectuée avec le relatlimab.
Nivolumab
Le blocage de la voie PD-1/PD-L1 a montré chez des modèles murins de grossesse une perturbation de la tolérance au fœtus et une augmentation de la perte fœtale. Les effets du nivolumab sur le développement prénatal et postnatal ont été évalués chez des singes ayant reçu du nivolumab 2 fois par semaine, du début de l'organogénèse, durant le premier trimestre, jusqu'à l'accouchement, à des niveaux d'exposition de 8 à 35 fois supérieurs à ceux observés à la dose clinique de nivolumab de 3 mg/kg (basés sur l'ASC). Il y a eu une augmentation dose-dépendante du nombre de pertes fœtales et de la mortalité néonatale à partir du troisième trimestre.
Le reste de la descendance des femelles traitées par nivolumab a survécu jusqu'au terme prévu, sans signes cliniques liés au traitement, ni anomalies du développement, ni altération du poids des organes ou modifications pathologiques macroscopiques ou microscopiques. Les résultats des courbes de croissance, ainsi que les données de tératogénicité, les données neurocomportementales et immunologiques, et les paramètres de pathologie clinique analysés pendant une période post-natale de 6 mois ont été comparables à ceux du groupe contrôle. Toutefois, sur la base du mécanisme d'action des deux molécules, l'exposition fœtale au nivolumab, et de la même manière, au relatlimab, peut augmenter le risque d'apparition de troubles d'origine immunologique ou altérer la réponse immunitaire normale. Par ailleurs, des troubles d'origine immunologique ont été rapportés chez des souris déficientes en PD-1 et PD-1/LAG-3. Aucune étude de fertilité n'a été réalisée avec le nivolumab.
Opdualag est fourni dans un flacon unidose et ne contient pas de conservateurs. La préparation doit être effectuée par du personnel formé, conformément aux règles de bonne pratique, notamment en matière d'asepsie.
Opdualag peut être utilisé en administration intraveineuse,
soit :
- sans
dilution, après transfert dans un récipient pour perfusion en utilisant une
seringue stérile appropriée ; soit
- après
dilution, conformément aux instructions suivantes :
- la
concentration finale de la perfusion doit être comprise entre 3 mg/mL et 12
mg/mL pour le nivolumab et entre 1 mg/mL et 4 mg/mL pour le relatlimab ;
- le volume total de la perfusion ne doit pas dépasser 160 mL. Pour les patients pesant moins de 40 kg, le volume total de la perfusion ne doit pas dépasser 4 mL par kilogramme de poids du patient.
La solution à diluer d'Opdualag peut être diluée avec :
- une
solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) ; ou
- une solution injectable de glucose à 50 mg/mL (5 %).
Préparation
de la perfusion
- Inspecter la solution à diluer d'Opdualag à la recherche de particules étrangères ou d'une coloration anormale. Ne pas secouer le flacon. Opdualag est une solution claire à opalescente, incolore à légèrement jaune. Jeter le flacon si la solution est trouble, présente une coloration anormale ou contient des particules étrangères.
- Prélever le volume nécessaire de solution à diluer d'Opdualag en utilisant une seringue stérile de volume approprié et transférer la solution à diluer dans un récipient pour perfusion stérile (éthylène-acétate de vinyle [EVA], polychlorure de vinyle [PVC] ou polyoléfine).
- Le cas échéant, diluer la solution d'Opdualag avec le volume nécessaire de solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) ou de solution injectable de glucose à 50 mg/mL (5 %). Afin de faciliter la préparation, la solution à diluer peut directement être transvasée dans une poche pré-remplie contenant le volume approprié de solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) ou de solution injectable de glucose à 50 mg/mL (5 %).
- Mélanger doucement la perfusion par rotation manuelle. Ne pas secouer.
Administration
La perfusion d'Opdualag ne doit être administrée ni en injection rapide ni en bolus intraveineux.
Administrer la perfusion d'Opdualag par voie intraveineuse sur une période de 30 minutes. Il est recommandé d'utiliser un set de perfusion et un filtre en ligne ou complémentaire, stérile, apyrogène, à faible liaison aux protéines (diamètre des pores compris entre 0,2 µm et 1,2 µm).
La perfusion d'Opdualag est compatible avec les poches en
EVA, en PVC et en polyoléfine, les sets de perfusion en PVC, et les filtres en
ligne avec membranes en polyéthersulfone (PES), en nylon et en polyfluorure de
vinylidène (PVDF), et diamètres de pores de 0,2 µm à 1,2 µm.
Ne pas administrer simultanément avec d'autres
médicaments sur la même ligne intraveineuse.
Après administration de la dose
d'Opdualag, rincer la ligne de perfusion avec une solution injectable de
chlorure de sodium à 9 mg/mL (0,9 %) ou une solution injectable de glucose à 50
mg/mL (5 %).
Élimination
Ne conserver aucune fraction inutilisée de la solution pour perfusion en vue de l'utiliser ultérieurement. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Médicament nécessitant une surveillance particulière pendant
le traitement.
Prescription réservée aux médecins compétents en
CANCEROLOGIE.
Prescription réservée aux spécialistes et services ONCOLOGIE
MEDICALE.
Réservé à l'usage HOSPITALIER.
Solution à diluer pour perfusion (solution stérile).
Liquide clair à opalescent, incolore à légèrement jaune, essentiellement exempt de particules. Le pH de la solution est approximativement de 5,8 et l'osmolalité approximativement de 310 mOsm/kg.
Boîte contenant un flacon de 25 mL (verre de type I) muni d'un bouchon (caoutchouc butyl enrobé) et d'un opercule amovible jaune en aluminium. Chaque flacon contient 21,3 mL de solution, soit un excédent de 1,3 mL.
Chaque mL de solution à diluer pour perfusion contient 12 mg de nivolumab et 4 mg de relatlimab. Un flacon de 20 mL contient 240 mg de nivolumab et 80 mg de relatlimab.
Le nivolumab et le relatlimab sont des anticorps monoclonaux humains de type immunoglobuline G4 (IgG4) produits sur des cellules ovariennes de hamster chinois, par la technologie de l'ADN recombinant.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Histidine
Chlorhydrate d'histidine monohydraté
Saccharose
Acide pentétique (acide
diéthylène-triamine-penta-acétique)
Polysorbate 80 (E433)
Eau pour préparations injectables