
VOYDEYA 50 mg 100 mg, comprimé pelliculé, boîte de 2 flacons de 90
Dernière révision : 19/04/2024
Taux de TVA : 2.1%
Laboratoire exploitant : ALEXION PHARMA FRANCE SAS
Source :

Voydeya en association avec le ravulizumab ou l'eculizumab est indiqué dans le traitement des patients adultes atteints d'hémoglobinurie paroxystique nocturne (HPN) présentant une anémie hémolytique résiduelle (voir rubrique Propriétés pharmacodynamiques).
-
Hypersensibilité à la substance active ou à l'un des excipients
mentionnés à la rubrique Liste des excipients.
-
Patients présentant une infection par Neisseria meningitidis non
résolue lors de l'instauration du traitement (voir rubrique Mises en garde spéciales et précautions d'emploi).
-
Patients n'étant pas vaccinés contre Neisseria meningitidis, à
moins qu'ils ne reçoivent une antibioprophylaxie appropriée jusqu'à deux
semaines après la vaccination (voir rubrique Mises en garde spéciales et précautions d'emploi).
Générales
Danicopan ne doit pas être administré en monothérapie car l'efficacité n'a pas été établie. Il doit être prescrit en association avec le ravulizumab ou l'eculizumab.
Infections graves
Infections à
méningocoque
Les patients traités par un inhibiteur du complément peuvent
présenter une prédisposition accrue aux infections à méningocoque (Neisseria
meningitidis). La vaccination antiméningococcique des patients conformément
aux recommandations vaccinales nationales en vigueur doit être à jour avant l'administration
de la première dose de danicopan.
Les patients chez lesquels le traitement est débuté moins de deux semaines après la vaccination antiméningococcique doivent recevoir une antibioprophylaxie appropriée pendant une durée allant jusqu'à deux semaines après la vaccination. Les patients doivent être vaccinés contre les sérogroupes A, C, Y et W135 afin de prévenir les infections par les sérogroupes méningococciques les plus couramment pathogènes. La vaccination contre le sérogroupe B, si le vaccin est disponible, est également recommandée. Il convient de tenir compte des recommandations officielles concernant l'utilisation appropriée des antibactériens.
Tous les patients traités par le danicopan doivent être surveillés afin que des signes précoces d'infection et de septicémie à méningocoque puissent être détectés, examinés immédiatement en cas de suspicion d'infection et traités par les antibiotiques appropriés. Les patients doivent être informés de ces signes et symptômes et de la conduite à tenir pour obtenir une prise en charge médicale immédiate.
Autres infections
graves
Le danicopan doit être administré avec prudence chez les
patients présentant une infection systémique active. Le danicopan bloque
sélectivement l'activation de la voie alterne du complément ; par conséquent,
les patients peuvent présenter une prédisposition accrue à d'autres infections
graves (autres que les infections à Neisseria meningitidis). Il est
recommandé que les patients soient vaccinés conformément aux recommandations
vaccinales en vigueur avant l'instauration du traitement par le danicopan en
association avec le ravulizumab ou l'eculizumab.
Insuffisance rénale sévère
Les patients atteints d'insuffisance rénale sévère chez lesquels la dose est augmentée à 150 mg trois fois par jour doivent être surveillés pour détecter tout événement indésirable pendant le traitement par le danicopan en raison d'une exposition plus élevée chez ces patients.
Poids faible
Les patients pesant moins de 60 kg doivent être surveillés pour détecter tout événement indésirable pendant le traitement par le danicopan en raison d'une exposition plus élevée attendue chez ces patients.
Augmentation des enzymes hépatiques
Des augmentations de l'alanine aminotransférase (ALAT) ont été observées dans les études cliniques (voir rubrique Effets indésirables). Un bilan hépatique est recommandé avant le début du traitement. Après l'instauration du traitement, une surveillance régulière des paramètres biochimiques conformément à la prise en charge de l'HPN est recommandée. L'interruption ou l'arrêt du traitement doivent être envisagés si les augmentations sont cliniquement significatives ou si les patients deviennent symptomatiques. Le danicopan n'est pas recommandé chez les patients présentant une insuffisance hépatique sévère (voir rubrique Posologie et mode d'administration).
Arrêt du traitement
Chez des volontaires sains, aux doses supérieures à 200 mg trois fois par jour, des augmentations de l'ALAT sont survenues après l'arrêt du traitement sans réduction progressive de la dose (voir rubrique Surdosage). En cas d'arrêt du traitement, la dose doit être réduite progressivement sur une période de 6 jours jusqu'à l'arrêt complet (voir rubrique Posologie et mode d'administration).j
Excipients à effet notoire :
Lactose
Ce médicament contient du lactose monohydraté. Les patients
présentant une intolérance au galactose, un déficit total en lactase ou un
syndrome de malabsorption du glucose et du galactose (maladies héréditaires
rares) ne doivent pas prendre ce médicament.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par
comprimé pelliculé, c'est-à-dire qu'il est essentiellement « sans sodium ».
Résumé du profil de sécurité
Les effets indésirables les plus fréquents sont : fièvre (25,0 %), céphalées (19,8 %), augmentations des enzymes hépatiques (11,5 %) et douleur dans les membres (11,5 %).
Tableau des effets indésirables
Le tableau 1 présente les effets indésirables rapportés dans les études cliniques du danicopan. Les effets indésirables sont présentés par classe de systèmes d'organes et terme préférentiel selon la convention de fréquence MedDRA : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10) et peu fréquent (≥ 1/1 000, < 1/100). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau 1 : Liste des effets indésirables
|
Classe de système d'organes MedDRA |
Très fréquent (≥ 1/10) |
Fréquent (≥ 1/100, < 1/10) |
|
Affections du système nerveux |
Céphalées |
|
|
Affections vasculaires |
|
Hypertension |
|
Affections gastrointestinales |
|
Vomissements |
|
Affections hépatobiliaires |
Augmentations des enzymes hépatiquesa |
|
|
Affections musculosquelettiques et du tissu conjonctif |
Douleur dans les membres |
|
|
Troubles généraux et anomalies au site d'administration |
Fièvre |
|
a Le terme « augmentations des enzymes hépatiques
» inclut les termes préférentiels augmentation de l'alanine aminotransférase
(ALAT), fonction hépatique anormale, augmentation des taux d'enzymes hépatiques
et augmentation des transaminases.
Description de certains effets indésirables
Augmentations des enzymes hépatiques
Pendant la période randomisée contrôlée de 12 semaines de l'étude ALXN2040-PNH-301, des anomalies des paramètres biologiques liées à des augmentations du taux d'ALAT ont été observées chez 14,0 % des patients traités par le danicopan. Chez les patients traités par le danicopan, des augmentations de l'ALAT > 3 × la limite supérieure de la normale (LSN) et ≤ 5 × LSN sont survenues chez 8,8 % des patients et des augmentations > 5 × LSN et ≤ 10 × LSN ont été observées chez 5,3 % des patients. Tous les patients étaient asymptomatiques et toutes les augmentations ont été transitoires. Certains cas d'augmentations sont survenus dans le cadre d'une hémolyse.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
Ne pas administrer en monothérapie; doit être prescrit en association avec le ravulizumab ou l'eculizumab.
AVANT INSTAURATION du traitement :
- La vaccination antiméningococcique conformément aux recommandations vaccinales nationales en vigueur doit être à jour.
Les patients chez lesquels le traitement est débuté moins de deux
semaines après la vaccination antiméningococcique doivent recevoir une
antibioprophylaxie appropriée pendant une durée allant jusqu'à deux
semaines après la vaccination.
- Il est recommandé que les patients soient vaccinés conformément aux recommandations vaccinales en vigueur.
- Réaliser un bilan hépatique.
SURVEILLANCE :
- Surveillance régulière des paramètres biochimiques conformément à la prise en charge de l'HPN.
INFORMER LA PATIENTE désirant allaiter que l'allaitement ne doit pas débuter moins de trois jours après l'arrêt du traitement.
Grossesse
Il n'existe pas de données sur l'utilisation du danicopan chez la femme enceinte. Les études effectuées chez l'animal n'ont pas mis en évidence d'effets délétères directs ou indirects sur la reproduction à la dose thérapeutique (voir rubrique Données de sécurité préclinique). Par mesure de précaution, il est préférable d'éviter l'utilisation de Voydeya pendant la grossesse.
Allaitement
Les données pharmacodynamiques/toxicologiques disponibles chez l'animal ont mis en évidence l'excrétion du danicopan/des métabolites dans le lait (voir rubrique Données de sécurité préclinique). Un risque pour les nouveau-nés/nourrissons ne peut être exclu. Voydeya ne doit pas être utilisé pendant l'allaitement et l'allaitement ne doit pas débuter moins de trois jours après l'arrêt du traitement.
Fertilité
Il n'existe pas de données sur l'effet du danicopan sur la fertilité humaine. Les études effectuées chez l'animal ont montré des effets possibles sur la fertilité masculine et sur l'efficacité de la reproduction (voir rubrique Données de sécurité préclinique).
Effets du danicopan sur d'autres médicaments
Substrats de la
P-gp (P-glycoprotéine)
L'administration concomitante par voie orale d'une dose
unique de 180 mg de fexofénadine, un substrat de la P-gp, avec le danicopan
(doses quotidiennes de 150 mg trois fois par jour) a entraîné des augmentations
de respectivement 1,42 fois et 1,62 fois de la Cmax et de l'ASC0-inf
de la fexofénadine. Les résultats semblent indiquer que le danicopan est un
inhibiteur faible de la P-gp. Des précautions peuvent être nécessaires en cas
d'administration concomitante de médicaments qui sont des substrats de la P-gp
(par exemple dabigatran, digoxine, edoxaban, fexofénadine, tacrolimus).
Substrats de la
BCRP (Breast Cancer Resistance Protein)
L'administration concomitante par voie orale d'une dose
unique de 20 mg de rosuvastatine, un substrat de la BCRP, avec le danicopan
(doses quotidiennes de 200 mg trois fois par jour) a entraîné des augmentations
de respectivement 3,29 fois et 2,25 fois de la Cmax et de l'ASC0-inf
de la rosuvastatine. Ce résultat semble indiquer que le danicopan est un
inhibiteur de la BCRP. Des précautions peuvent être nécessaires en cas
d'administration concomitante de médicaments qui sont des substrats de la BCRP
(par exemple rosuvastatine et sulfasalazine).
Le traitement doit être instauré par un médecin expérimenté dans la prise en charge des patients présentant des affections hématologiques.
Posologie
La dose initiale recommandée est de 150 mg prise par voie orale trois fois par jour, à environ 8 heures d'intervalle (± 2 heures). En fonction de la réponse clinique, la dose peut être augmentée à 200 mg trois fois par jour après au moins quatre semaines de traitement
Oubli d'une dose
En cas d'oubli d'une dose, les patients doivent prendre la
dose oubliée dès qu'ils s'en rendent compte, sauf s'il est presque l'heure de
prendre la dose suivante ; dans ce cas, ils ne doivent pas prendre la dose
oubliée, mais prendre la dose suivante au moment habituel. Les patients doivent
être informés qu'ils ne doivent pas prendre deux doses ou plus en même temps.
Arrêt du
traitement
Du fait de la possibilité d'augmentations de l'alanine
aminotransférase (ALAT) après l'arrêt du traitement (voir rubrique Mises en garde spéciales et précautions d'emploi), en cas
d'arrêt du traitement, la dose doit être réduite progressivement sur une
période de 6 jours jusqu'à l'arrêt complet, comme suit :
-
Dose de 100 mg : 100 mg deux fois par jour pendant 3 jours, puis 100 mg une
fois par jour pendant 3 jours.
-
Dose de 150 mg : 100 mg trois fois par jour pendant 3 jours, puis 50 mg
trois fois par jour pendant 3 jours.
-
Dose de 200 mg : 100 mg trois fois par jour pendant 3 jours, puis 100 mg
deux fois par jour pendant 3 jours.
Populations particulières
Sujets âgés
Aucun ajustement de la posologie n'est nécessaire chez les
patients âgés. Cependant, l'expérience du danicopan chez les patients âgés de
65 ans et plus est limitée (voir rubrique Propriétés pharmacodynamiques).
Insuffisance
rénale
Aucun ajustement de la posologie n'est nécessaire chez les
patients présentant une insuffisance rénale légère (débit de filtration
glomérulaire estimé [DFGe] ≥ 60 et < 90 mL/min/1,73 m2) ou
modérée (DFGe ≥ 30 et < 60 mL/min/1,73 m2). Chez les
patients présentant une insuffisance rénale sévère (DFGe < 30 mL/min/1,73 m2),
la dose initiale recommandée est de 100 mg trois fois par jour par voie orale,
prise à intervalle d'environ 8 heures (± 2 heures). En fonction de la réponse
clinique, la dose peut être augmentée à 150 mg trois fois par jour après au
moins quatre semaines de traitement (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacocinétiques).
Insuffisance
hépatique
Aucun ajustement de la posologie n'est nécessaire chez les
patients présentant une insuffisance hépatique légère (classe A de Child-Pugh)
à modérée (classe B de Child-Pugh) (voir rubrique Propriétés pharmacocinétiques). Il n'a pas été mené
d'études chez les patients atteints d'insuffisance hépatique sévère (classe C
de Child-Pugh). Par conséquent, l'utilisation du danicopan n'est pas recommandée
dans cette population (voir rubrique Mises en garde spéciales et précautions d'emploi).
Population pédiatrique
La sécurité et l'efficacité de Voydeya chez les enfants et adolescents âgés de moins de 18 ans n'ont pas encore été établies. Aucune donnée n'est disponible.
Mode d'administration
Voie orale.
Les comprimés doivent être pris avec un repas ou une
collation (voir rubrique Propriétés pharmacocinétiques).
Durée de conservation :
30 mois dans le flacon en polyéthylène haute densité (PEHD)
Après première ouverture du flacon : 48 jours
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
Des doses uniques allant jusqu'à 1 200 mg et des doses répétées allant jusqu'à 800 mg deux fois par jour ont été administrées à des volontaires sains. Des augmentations de l'ALAT sont survenues après l'arrêt du traitement sans réduction progressive de la dose chez deux sujets qui avaient reçu les doses de 500 mg et 800 mg deux fois par jour pendant 14 jours. Toutes les anomalies des taux d'ALAT ont été transitoires, sans signes d'anomalies de la fonction hépatique, et se sont résolues spontanément.
En cas de surdosage, des augmentations de l'alanine aminotransférase et d'autres paramètres hépatiques peuvent survenir. Les mesures de support générales sont recommandées. On ne sait pas si le danicopan peut être éliminé par la dialyse.
Classe pharmacothérapeutique : Immunosuppresseurs, Inhibiteurs du complément, Code ATC : L04AJ09
Mécanisme d'action
Le danicopan se lie de façon réversible au facteur D du complément et agit comme un inhibiteur sélectif de la fonction de ce facteur. En inhibant le facteur D, le danicopan bloque sélectivement l'activation de la voie alterne du complément, ce qui empêche la synthèse de plusieurs protéines effectrices, dont les fragments de C3, après l'activation de la voie alterne. Les deux autres voies du complément (voie classique et voie des lectines) restent actives. L'effet inhibiteur du danicopan sur l'activation de la voie alterne inhibe le dépôt de fragments de C3 à la surface des érythrocytes HPN ; ce dépôt est une cause majeure de l'hémolyse extra-vasculaire (HEV) qui peut devenir cliniquement significative dans un petit sous-groupe de patients atteints d'HPN traités par un inhibiteur de C5. Le maintien de l'inhibition de la protéine C5 permet de contrôler les conséquences physiopathologiques engageant le pronostic vital de l'activation de la voie terminale du complément sous-jacente à l'HPN.
Effets pharmacodynamiques
Dans une étude clinique menée chez des patients atteints d'HPN présentant une HEV cliniquement significative traités par le ravulizumab ou l'eculizumab, le danicopan a induit l'inhibition attendue de l'activité de la voie alterne, une réduction des taux plasmatiques de Bb (un produit de clivage du facteur B du complément par le facteur D), ainsi qu'une diminution du dépôt de fragments de C3 sur les érythrocytes HPN circulants.
Électrophysiologie
cardiaque
Des doses uniques de 400 mg, 800 mg et 1 200 mg de danicopan
administrées par voie orale n'ont pas entraîné d'allongement de l'intervalle
QTc. Il n'a pas été observé de signaux explicites concernant des anomalies des
intervalles ou des formes d'ondes sur l'ECG.
Efficacité et sécurité cliniques
L'efficacité et la sécurité du danicopan chez les patients adultes atteints d'HPN présentant une HEV cliniquement significative ont été évaluées dans une étude de phase III internationale, randomisée en double aveugle, contrôlée contre placebo (étude ALXN2040PNH301). Dans l'étude ont été inclus 86 patients atteints d'HPN qui avaient été traités par le ravulizumab ou l'eculizumab à une dose stable pendant au moins les six mois précédant l'inclusion et qui présentaient une anémie (taux d'hémoglobine [Hb] ≤ 9,5 g/dL [5,9 mmol/L]) avec un taux de réticulocytes ≥ 120 × 109/L, avec ou sans support transfusionnel.
Le danicopan était administré conformément à la posologie recommandée indiquée à la rubrique Posologie et mode d'administration (150 mg trois fois par jour, et jusqu'à une dose maximale de 200 mg trois fois par jour en fonction de la réponse clinique).
Les antécédents vaccinaux étaient passés en revue et les patients devaient recevoir une vaccination antiméningococcique avant ou au moment de l'instauration du traitement par le danicopan si le statut vaccinal au cours des trois ans précédents ne pouvait pas être vérifié.
Les patients ont été randomisés selon un rapport 2:1 pour recevoir le danicopan ou le placebo trois fois par jour pendant 12 semaines en plus du traitement de fond par ravulizumab ou eculizumab administré dans les deux groupes. Après la semaine 12, tous les patients ont reçu le danicopan en association avec leur traitement de fond par ravulizumab ou eculizumab jusqu'à la semaine 24. À la fin des périodes de traitement (semaine 24), il était proposé aux patients d'entrer dans une période d'extension à longterme au cours de laquelle ils continuaient à recevoir le danicopan avec le traitement de fond par ravulizumab ou eculizumab.
Les caractéristiques démographiques ou initiales étaient généralement équilibrées entre les groupes de traitement. L'histoire de l'HPN était comparable entre le groupe recevant le traitement actif et le groupe contrôle recevant le placebo. L'âge moyen à l'inclusion était de 52,8 ans et la majorité des patients étaient de sexe féminin (62,8 %). Lors de l'inclusion, le taux moyen d'hémoglobine était de 7,75 g/dL [4,81 mmol/L] et le taux moyen de réticulocytes était de 239,40 × 109/L. Au cours des 24 semaines précédant l'administration de la première dose, 76 patients (88,4 %) avaient reçu des transfusions de concentré érythrocytaire ou de sang total et le nombre moyen de transfusions était de 2,6. Le taux moyen de LDH était de 298,13 U/L et le score FACIT-Fatigue moyen de 33,24. Dans l'étude ont été inclus 51 patients (59,3 %) traités par ravulizumab et 35 patients (40,7 %) traités par eculizumab.
Le critère d'évaluation principal était la variation du taux d'Hb entre l'inclusion et la semaine 12. Les critères secondaires étaient le pourcentage de patients ayant obtenu une augmentation ≥ 2 g/dL [1,2 mmol/L] du taux d'Hb à la semaine 12 en l'absence de transfusion, le pourcentage de patients n'ayant pas eu besoin de transfusion (absence de recours aux transfusions) jusqu'à la semaine 12, la variation du score du questionnaire d'évaluation fonctionnelle de la fatigue dans le traitement des maladies chroniques (Functional Assessment of Chronic Illness Therapy [FACIT]-Fatigue) à la semaine 12 par rapport au score initial et la variation en nombre absolu des réticulocytes à la semaine 12 par rapport à la valeur initiale. L'absence de recours aux transfusions était considérée comme atteint uniquement chez les patients qui n'avaient pas reçu de transfusions et pour lesquels aucune transfusion n'était justifiée selon les recommandations du protocole entre l'inclusion et la semaine 12 de la première période de traitement.
Pour l'analyse de l'efficacité, les données principales sont fondées sur une analyse prédéfinie effectuée après que les 63 premiers patients randomisés aient atteint la fin de la première période de traitement de 12 semaines (traitement terminé ou arrêté). Le danicopan en association avec le ravulizumab ou l'eculizumab a été supérieur au placebo en association avec le ravulizumab ou l'eculizumab pour le critère d'évaluation principal et a induit une augmentation statistiquement significative du taux d'Hb entre l'inclusion et la semaine 12. La variation de la moyenne des moindres carrés (MC) du taux d'hémoglobine par rapport à la valeur initiale était de 2,94 g/dL [1,82 mmol/L] dans le groupe danicopan contre 0,50 g/dL [0,31 mmol/L] dans le groupe placebo. La différence entre les groupes de traitement était de 2,44 g/dL [1,51 mmol/L] (IC à 95 % : 1,69 [1,05] ; 3,20 [1,99]) ; p < 0,0001). Par rapport au placebo, on observe avec danicopan une amélioration statistiquement significative des quatre critères d'évaluation secondaires : le pourcentage de patients ayant obtenu une augmentation ≥ 2 g/dL [1,2 mmol/L] du taux d'hémoglobine en l'absence de transfusion (59,5 % contre 0 %, différence entre les traitements : 46,9 [IC à 95 % : 29,2 ; 64,7] ; p < 0,0001), le pourcentage de patients n'ayant pas eu besoin de transfusion (83,3 % contre 38,1 %, différence entre les traitements : 41,7 [IC à 95 % : 22,7 ; 60,8] ; p = 0,0004), la variation du score FACIT Fatigue (7,97 contre 1,85, différence entre les traitements : 6,12 [IC à 95 % : 2,33 ; 9,91] ; p = 0,0021) et la variation du taux de réticulocytes (-83,8 contre 3,5, différence entre les traitements : -87,2 [IC à 95 % : -117,7 ; -56,7] ; p < 0,0001).
Les résultats supplémentaires à la semaine 12 chez l'ensemble des patients randomisés (N = 86), concordent avec les résultats issus de l'analyse principale de l'efficacité (N = 63). Le danicopan en association avec le ravulizumab ou l'eculizumab a été supérieur au placebo en association avec le ravulizumab ou l'eculizumab pour le critère d'évaluation principal et a induit une augmentation statistiquement significative du taux d'Hb entre l'inclusion et la semaine 12 (voir tableau 2 et figure 1). Par rapport au placebo, le danicopan a également induit des améliorations statistiquement significatives pour les quatre critères d'évaluation secondaires (voir tableau 2).
Pendant la première période de traitement de 12 semaines, la dose a été augmentée de 150 mg à 200 mg trois fois par jour chez 14 des 57 patients (24,6 %) du groupe danicopan en association. Quatre patients (deux patients randomisés dans le groupe placebo et deux patients randomisés dans le groupe danicopan) ont arrêté le traitement pendant la première période de traitement. Il n'a pas été rapporté d'arrêts du traitement en raison d'une hémolyse.
Tableau 2 : Analyses des résultats à la semaine 12 du critère d'évaluation principal et des critères secondaires (ensemble des patients randomisés)
|
|
Danicopan (en association avec le ravulizumab ou l'eculizumab) N = 57 |
Placebo (en association avec le ravulizumab ou l'eculizumab) N = 29 |
|
Variation du taux d'hémoglobine (critère principal) |
||
|
Variation moyenne entre l'inclusion et la semaine 12 (g/dL [mmol/L]) |
2,81 [1,74] |
0,46 [0,29] |
|
Différence entre les traitements* (IC à 95 %) |
2,35 [1,46] (1,63 [1,01] ; 3,06 [1,90]) |
|
|
Pourcentage de patients ayant obtenu une augmentation ≥
2 g/dL [1,2 mmol/L] du taux d'hémoglobine en l'absence de transfusion |
||
|
À la semaine 12 (%) |
54,4 |
0 |
|
Différence entre les traitements** (IC à 95 %) |
47,5 (32,6 ; 62,4) |
|
|
Pourcentage de patients n'ayant pas eu besoin de transfusion |
||
|
Jusqu'à la semaine 12 de la période de traitement (%) |
78,9 |
27,6 |
|
Différence entre les traitements**(IC à 95 %) |
48,4 (31,8 ; 64,9) |
|
|
Variation du score FACIT Fatigue |
||
|
Variation moyenne entre l'inclusion et la semaine 12 |
8,10 |
2,38 |
|
Différence entre les traitements* (IC à 95 % ) |
5,72 (2,62 ; 8,83) |
|
|
Variation du taux de réticulocytes |
||
|
Variation moyenne entre l'inclusion et la semaine 12 (109/L) |
-92,6 |
-0,9 |
|
Différence entre les traitements* (IC à 95 %) |
-91,6 (-120,0 ; -63,3) |
|
* Selon un modèle à effets mixtes pour mesures répétées.
** Les différences des taux et les IC à 95 % associés ont été calculés selon la méthode de Miettinen et Nurminen avec ajustement pour les facteurs de stratification.
** Les différences des taux et les IC à 95 % associés ont été calculés selon la méthode de Miettinen et Nurminen avec ajustement pour les facteurs de stratification.
Abréviations : IC = intervalle de confiance ; FACIT = Functional Assessment of Chronic Illness Therapy.
Figure 1 : Variation moyenne du taux d'hémoglobine entre l'inclusion et la semaine 12 (ensemble des patients randomisés)
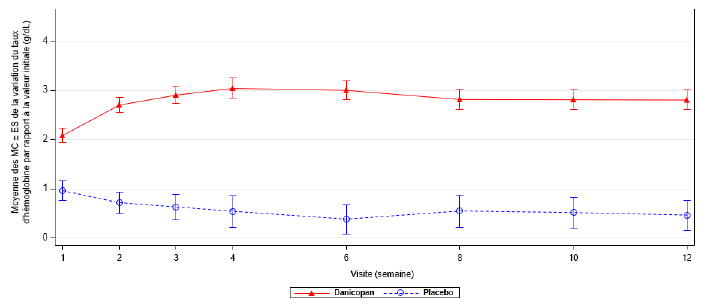
Les résultats à la semaine 24 concordaient avec ceux observés à la semaine 12, ce qui corrobore le maintien de l'effet. Chez les 55 patients atteints d'HPN traités par le danicopan pendant 24 semaines, la variation de la moyenne des MC (Moyenne Cumulative) du taux d'Hb entre l'inclusion et la semaine 24 était de 2,95 g/dL [1,83 mmol/L] (IC à 95 % : 2,42 [1,50] ; 3,48 [2,16]), l'absence de recours aux transfusions a été maintenue jusqu'à la semaine 24 chez 69,1 % des patients, et 41,8 % des patients avaient obtenu une augmentation ≥ 2 g/dL [1,2 mmol/L] du taux d'Hb à la semaine 24 en l'absence de transfusion. Il a également été observé chez ces patients des améliorations uniformes du score FACIT-Fatigue qui ont été maintenues pendant 24 semaines ; la variation moyenne par rapport au score initial était de 6,19 points (IC à 95 % : 4,10 ; 8,29).
Les résultats d'efficacité jusqu'à la semaine 72 concordent avec ceux observés aux semaines 12 et 24 et confirment la durabilité et le maintien de l'effet au cours du temps. Chez les patients ayant reçu le danicopan pendant 72 semaines (N = 16), la variation moyenne du taux d'Hb entre l'inclusion et la semaine 72 était de 2,99 g/dL [1,86 mmol/L].
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec Voydeya dans un ou plusieurs sous-groupes de la population pédiatrique dans l'indication d'HPN (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Absorption
Après administration par voie orale, le danicopan est absorbé rapidement, le temps jusqu'à la concentration maximale observée (Tmax) moyen étant d'environ 3 heures. Au-delà de la dose comprise entre 200 mg et 800 mg, la Cmax augmentait de façon moins proportionnelle à la dose, probablement en raison de la faible solubilité limitant l'absorption. Après administration de danicopan avec un repas à haute teneur en lipides, l'ASC et la Cmax étaient plus élevées d'environ 25 % et 93 % respectivement par rapport à l'administration à jeun. Le Tmax médian était comparable lorsque le danicopan était administré avec un repas ou à jeun, et d'environ 3,0 et 2,5 heures respectivement (voir rubrique Posologie et mode d'administration).
Le danicopan présente une capacité de pénétration tissulaire élevée et est un substrat de la P-gp in vitro, mais à faible ratio d'efflux. L'exposition au danicopan après administration orale ne semble pas être modifiée par l'efflux induit par la P-gp dans le tractus gastro-intestinal. Le danicopan n'est pas un substrat de la BCRP, d'OATP1B1 ni d'OATP1B3.
Distribution
Le danicopan est fortement lié aux protéines plasmatiques humaines (91,5 % à 94,3 %) et est distribué principalement dans le plasma, avec un rapport des ASC0-∞ sang total/plasma moyennes de 0,545. Les concentrations plasmatiques du danicopan semblaient diminuer de façon biphasique après l'atteinte du Tmax. Après administration par voie orale, le volume apparent de distribution chez un patient de 75 kg estimé selon le modèle de pharmacocinétique de population était de 168 L pour le compartiment central et de 234 L pour le compartiment périphérique (402 L au total), ce qui semble indiquer une distribution modérée du danicopan dans le compartiment périphérique.
Biotransformation
Après administration par voie orale, le danicopan est fortement métabolisé (96 %) par des voies d'oxydation, de réduction et d'hydrolyse, l'hydrolyse du groupe amide étant identifiée comme la voie d'élimination majeure. Le métabolisme catalysé par les enzymes du CYP450 est minime.
Élimination
Après administration par voie orale, le danicopan est éliminé principalement dans les fèces (environ
69 % de la dose administrée contre environ 25 % dans les urines). Dans l'analyse pharmacocinétique (PK) de population portant sur des patients atteints d'HPN présentant une hémolyse extra-vasculaire (HEV) cliniquement significative, la valeur moyenne estimée du t1/2 était de 7,91 heures.
Populations particulières
L'analyse PK de population n'a pas montré de différences significatives de la pharmacocinétique du danicopan en fonction du sexe, de l'âge ou du groupe ethnique.
Insuffisance
rénale
Après administration par voie orale de 200 mg de danicopan
chez des sujets présentant une insuffisance rénale sévère (DFGe < 30
mL/min/1,73 m2), l'exposition (ASC) au danicopan était augmentée
d'environ 50 % par rapport à l'exposition chez les sujets ayant une fonction
rénale normale. L'excrétion rénale n'est pas la voie d'élimination majeure du
danicopan, même chez les sujets ayant une fonction rénale normale (voir
rubrique Posologie et mode d'administration).
Insuffisance
hépatique
Il n'a pas été observé de différence significative de
l'exposition au danicopan chez les sujets présentant une insuffisance hépatique
modérée (classe B de Child-Pugh) par rapport aux sujets ayant une fonction
hépatique normale (voir rubrique Posologie et mode d'administration). Il n'a pas été mené d'études chez les
patients présentant une insuffisance hépatique sévère (classe C de Child-Pugh).
Voydeya n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Dans l'étude de toxicologie de 6 mois chez le rat (espèce non sensible aux effets pharmacologiques du danicopan), une hypertrophie du foie, de la thyroïde et des glandes surrénales ont été observées à la dose de 1 000 mg/kg/jour, dose sans effet nocif observé (DSENO) environ 26 fois supérieure à l'exposition humaine à la dose de 200 mg trois fois par jour sur la base de l'ASC).
Dans l'étude de toxicologie de 9 mois chez le chien, la dose de 150 mg/kg/jour n'a pas été tolérée. Des effets sur les organes cibles ont été observés dans le foie, compatibles avec une cholestase hépatobiliaire, et comprenaient une hypertrophie/hyperplasie des voies biliaires et une accumulation de pigments biliaires dans les cellules de Kupffer et les hépatocytes. Les augmentations des taux d'ASAT, d'ALAT, de phosphatase alcaline, de gamma-glutamyltransférase et de bilirubine totale étaient corrélées aux anomalies histologiques dans le foie. L'hypertrophie/hyperplasie des voies biliaires a été observée chez les mâles aux doses ≥ 75 mg/kg/jour (environ 5 fois supérieure à l'exposition humaine à la dose de 200 mg trois fois par jour sur la base de l'ASC).
Cependant, à une dose de 75 mg/kg/jour, la sévérité et l'ampleur des anomalies étaient moindres et ces observations n'étaient pas corrélées avec les résultats de pathologie clinique.
Génotoxicité/carcinogénicité
Le danicopan n'a pas été génotoxique dans l'essai de mutation réverse sur bactéries (test d'Ames), dans le test des micronoyaux in vitro sur lymphocytes de sang périphérique humains ou dans le test des micronoyaux in vivo chez le rat.
Le danicopan n'a pas été cancérogène dans l'étude de carcinogénicité de 6 mois chez les souris TgRasH2 et dans l'étude de carcinogénicité de 2 ans chez les rat. Dans l'étude chez les rats toutefois, il a été observé une incidence élevée de néoplasies épithéliales de l'endomètre chez les animaux recevant la dose la plus élevée de 500 mg/kg/jour par rapport aux animaux témoins ; cependant, l'incidence générale des carcinomes endométriaux peut être élevée dans cette souche de rat. La pertinence clinique de ces observations n'est pas connue.
Toxicité sur la fertilité/sur le développement
Dans l'étude de la fertilité et du développement embryonnaire précoce chez le lapin, il a été observé une réduction des performances de reproduction chez les mâles et les femelles à la dose de 500 mg/kg/jour, une dose associée à une tolérance médiocre. La DSENO pour les fonctions de reproduction mâles et femelles estimée était de 250 mg/kg/jour (induisant une exposition représentant environ 7,2 fois et 8,8 fois l'exposition chez l'homme).
Dans l'étude du développement pré- et postnatal chez le lapin, chez les mâles de la génération F1, il a été constaté par rapport aux valeurs chez les animaux témoins une diminution de la concentration en sperme dans l'épididyme (de 19 %, 20 % et 18 %) dans tous les groupes de dose (50, 125 et 250 mg/kg/jour respectivement), la différence n'étant statistiquement significative que dans les groupes de dose faible et intermédiaire. Cela n'a pas eu d'effet sur les capacités de reproduction des mâles de la génération F1.
Chez le lapin, il n'a pas été observé d'effets sur le développement embryonnaire précoce et sur le développement fœtal ni sur le développement postnatal à une exposition systémique maternelle moyenne représentant environ 20 fois l'exposition chez l'homme. Chez le rat, il n'a pas été constaté d'effets sur le développement embryonnaire et fœtal à une exposition maternelle représentant jusqu'à 30 fois environ l'exposition chez l'homme à la dose de 200 mg trois fois par jour.
Excrétion dans le lait
Après administration par voie orale à des lapines du jour 4 au jour 10 de l'allaitement, le danicopan était excrété dans le lait, les concentrations dans le lait étant supérieures d'environ 5 fois et 3,5 fois aux concentrations plasmatiques maternelles aux doses de 50 et 250 mg/kg/jour respectivement.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Médicament nécessitant une surveillance particulière pendant
le traitement.
Prescription hospitalière.
Prescription réservée aux médecins compétents en maladie du
sang.
Prescription réservée aux spécialistes et services
HEMATOLOGIE.
Prescription réservée aux spécialistes et services MEDECINE
INTERNE.
Comprimé pelliculé.
Voydeya 50 mg comprimés pelliculés
Comprimés pelliculés ronds de couleur blanche à blanc cassé, portant les mentions « DCN » au-dessus de « 50 » gravées en creux sur une face et unis sur l'autre face. Le diamètre du comprimé est d'environ 8 mm.
Voydeya 100 mg comprimés pelliculés
Comprimés pelliculés ronds de couleur blanche à blanc cassé, portant les mentions « DCN » au-dessus de « 100 » gravées en creux sur une face et unis sur l'autre face. Le diamètre du comprimé est d'environ 10,3 mm.
Flacon
Flacons en PEHD avec fermeture de sécurité enfant et sachet déshydratant contenant 90 comprimés pelliculés. Chaque boîte contient 180 comprimés pelliculés.
Présentation :
- Boîte contenant un flacon de 90 comprimés pelliculés de 50 mg et un flacon de 90 comprimés pelliculés de 100 mg.
Voydeya 50 mg comprimés pelliculés
Chaque comprimé pelliculé contient 50 mg de danicopan.
Voydeya 100 mg comprimés pelliculés
Chaque comprimé pelliculé contient 100 mg de danicopan.
Excipient à effet notoire :
Chaque comprimé de 50 mg contient 57,5 mg de lactose
monohydraté.
Chaque comprimé de 100 mg contient 115 mg de lactose
monohydraté.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Noyau du comprimé
Lactose monohydraté
Cellulose microcristalline
Croscarmellose sodique
Laurilsulfate de sodium
Stéarate de magnésium
Silice colloïdale hydrophobe
Succinate d'acétate d'hypromellose
Pelliculage
Alcool polyvinylique
Dioxyde de titane (E171)
Macrogol 4000
Talc