
Eylea 114,3mg-ml solution injectable en seringue préremplie, boîte de 1 seringue préremplie de 0,184 mL
Dernière révision : 03/10/2024
Taux de TVA : 2.1%
Prix de vente : 657,61 €
Taux remboursement SS : 100%
Base remboursement SS : 657,61 €
Laboratoire exploitant : BAYER HEALTHCARE SAS
Source :

Eylea est indiqué chez l'adulte dans le traitement de :
-
la forme néovasculaire (humide) de la dégénérescence maculaire liée à
l'âge (DMLAn) (voir rubrique Propriétés pharmacodynamiques),
-
la baisse d'acuité visuelle due à l'œdème maculaire diabétique (OMD)
(voir rubrique Propriétés pharmacodynamiques).
- Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
- Infection oculaire ou périoculaire.
- Inflammation intraoculaire sévère active.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Réactions liées aux injections intravitréennes
Les injections intravitréennes, y compris celles d'Eylea, ont été associées à des endophtalmies, des inflammations intraoculaires, des décollements de la rétine, des déchirures de la rétine et des cataractes traumatiques (voir rubrique Effets indésirables). Des techniques d'injection aseptiques appropriées doivent toujours être utilisées lors de l'administration d'Eylea. Les patients doivent être informés que tout symptôme évocateur d'une endophtalmie ou de l'un des événements mentionnés ci-dessus doit être signalé sans délai et faire l'objet d'une prise en charge adaptée.
Augmentation de la pression intraoculaire
Des augmentations transitoires de la pression intraoculaire ont été observées dans les 60 minutes suivant des injections intravitréennes, y compris après injection d'Eylea (voir rubrique Effets indésirables). La pression intraoculaire ainsi que la perfusion de la tête du nerf optique doivent donc être surveillées et prises en charge de manière appropriée. Des précautions particulières sont nécessaires chez les patients présentant un glaucome mal contrôlé (ne pas injecter Eylea tant que la pression intraoculaire est ≥ 30 mmHg).
Immunogénicité
L'aflibercept étant une protéine thérapeutique, il existe un risque d'immunogénicité (voir rubrique Propriétés pharmacodynamiques). Les patients doivent être informés que tout signe ou symptôme d'inflammation intraoculaire doit être signalé, en particulier une douleur, une photophobie, ou une rougeur, qui peuvent être des signes cliniques liés à une hypersensibilité.
Effets systémiques
Des effets indésirables systémiques incluant des événements hémorragiques non oculaires et des événements thromboemboliques artériels ont été rapportés après injection intravitréenne d'inhibiteurs du VEGF. Il existe un risque théorique que ces événements soient liés à l'inhibition du VEGF (voir rubrique Effets indésirables).
Les données concernant la sécurité du traitement sont limitées chez les patients présentant une DMLAn ou un OMD et ayant des antécédents d'accident vasculaire cérébral, d'accident ischémique transitoire ou d'infarctus du myocarde dans les 6 derniers mois. La prudence s'impose lors du traitement de ces patients.
Traitement bilatéral
La sécurité et l'efficacité d'un traitement bilatéral impliquant l'injection d'Eylea 114,3 mg/mL dans chaque œil n'ont pas été étudiées (voir rubrique Propriétés pharmacodynamiques). Un traitement bilatéral pourrait entraîner une augmentation de l'exposition systémique et donc un risque accru d'effets indésirables systémiques.
Utilisation en association avec d'autres anti-VEGF
Les données disponibles sont limitées concernant l'utilisation d'Eylea en association avec d'autres médicaments anti-VEGF (administrés par voie systémique ou oculaire).
Suspension du traitement
Le traitement doit être suspendu en cas de :
- diminution de la meilleure acuité visuelle corrigée (MAVC) ≥ 30 lettres par rapport à la dernière évaluation de l'acuité visuelle ;
- décollement de la rétine rhegmatogène ou des trous maculaires de stade 3 ou 4 ;
- déchirure rétinienne ;
- hémorragie sous-rétinienne impliquant le centre de la fovéa ou lorsque la taille de l'hémorragie est ≥ 50 % de la surface totale de la lésion ;
- chirurgie intraoculaire réalisée dans les 28 jours précédents ou prévue dans les 28 jours suivants.
Déchirure de l'épithélium pigmentaire rétinien
Les facteurs de risque associés au développement d'une déchirure de l'épithélium pigmentaire rétinien lors du traitement de la DMLAn par un agent anti-VEGF incluent un décollement étendu et/ou profond de l'épithélium pigmentaire rétinien. La prudence est nécessaire lors de l'instauration du traitement par l'aflibercept chez les patients présentant ces facteurs de risque de déchirure de l'épithélium pigmentaire rétinien.
Femmes en âge de procréer
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement et pendant au moins 4 mois après la dernière injection intravitréenne d'Eylea 114,3 mg/mL (voir rubrique Fertilité, grossesse et allaitement).
Populations chez lesquelles les données sont limitées
Les données concernant le traitement par Eylea chez les patients diabétiques dont le taux d'HbA1c est supérieur à 12 %, ou les patients présentant une rétinopathie diabétique proliférante, sont limitées.
Eylea n'a pas été étudié chez les patients présentant une infection systémique active, ou une pathologie oculaire associée comme un décollement de la rétine ou un trou maculaire. Il n'existe pas non plus de données concernant le traitement par Eylea chez les patients diabétiques présentant une hypertension non contrôlée. Ce manque de données doit être pris en considération par le médecin au moment de traiter ces patients.
Résumé du profil de tolérance
Les effets indésirables graves étaient la cataracte (8,2 %), l'hémorragie rétinienne (3,6 %), l'augmentation de la pression intraoculaire (2,8 %), l'hémorragie vitréenne (1,2 %), la cataracte sous-capsulaire (0,95 %), la cataracte nucléaire (0,6%), le décollement de la rétine (0,6 %) et la déchirure rétinienne (0,5 %).
Les effets indésirables les plus fréquemment observés chez les patients traités par Eylea 114,3 mg/mL, étaient la cataracte (8,2 %), la baisse de l'acuité visuelle (4,4 %), les corps flottants vitréens (4,0 %), , l'hémorragie conjonctivale (3,8 %), le décollement du vitré (3,7 %), l'hémorragie rétinienne (3,6 %), l'augmentation de la pression intraoculaire (2,8 %), et douleur oculaire (2,0%).
Le profil de tolérance observé dans les 3 études cliniques a été similaire chez les patients traités par Eylea 114,3 mg/mL (N = 1 217) et par Eylea 40 mg/mL (N = 556) chez les patients présentant une DMLAn et un OMD.
Liste tabulée des effets indésirables
Au total, 1 217 patients traités par Eylea 114,3 mg/mL ont
constitué la population sélectionnée pour l'évaluation du profil de tolérance
dans 3 études cliniques de phase II/III (CANDELA, PULSAR, PHOTON).
Les données de sécurité décrites ci-dessous incluent tous
les effets indésirables rapportés avec une imputabilité possiblement liée à la
procédure d'injection ou au médicament.
Les effets indésirables sont présentés par classe de système
ou d'organe et par fréquence selon les règles suivantes : Très fréquent (≥
1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100),
rare (≥
1/10 000, < 1/1 000).
Dans chaque groupe de fréquence, les effets indésirables
sont présentés par ordre décroissant de gravité.
Tableau 1 : Effets indésirables survenus au cours du traitement pendant les études de phase II/III chez les patients atteints de DMLAn ou d'OMD et traités par Eylea 114,3 mg/mL
|
Classe de système ou d'organes |
Fréquent |
Peu fréquent |
Rare |
|
Affections du système immunitaire |
Hypersensibilité* |
|
|
|
Affections oculaires |
Cataracte, Augmentation de la pression intraoculaire, Corps flottants vitréens, Décollement du vitré, Hémorragie du vitré, Hémorragie rétinienne, Baisse de l'acuité visuelle, Douleur oculaire, Hémorragie conjonctivale, Kératite ponctuée Abrasion de la cornée |
Décollement de la rétine, Déchirure de la rétine, Déchirure de l'épithélium pigmentaire rétinien, Décollement de l'épithélium pigmentaire rétinien, Uvéite, Inflammation de l'iris, Iridocyclite, Inflammation du vitré, Cataracte corticale, Cataracte nucléaire, Cataracte sous-capsulaire, Érosion de la cornée, Vision trouble, Douleur au site
d'injection, Sensation de corps étrangers dans les yeux, Augmentation de la sécrétion lacrymale, Hémorragie au site d'injection, Hyperhémie conjonctivale, Œdème palpébral, Hyperhémie oculaire, Irritation au site d'injection |
Œdème cornéen, Opacifications du cristallin, Dégénérescence de la rétine, Irritation palpébrale |
Les effets indésirables suivants d'Eylea 40 mg/mL sont également considérés comme possibles avec Eylea 114,3 mg/mL mais n'ont pas été rapportés au cours des études cliniques menées avec Eylea 114,3 mg/mL : sensation intraoculaire anormale, défaut de l'épithélium cornéen, inflammation de la chambre antérieure, endophtalmie, cécité, cataracte traumatique, hypopyon, réactions anaphylactiques/anaphylactoïdes sévères.
Description de certains effets indésirables
Effets indésirables liés à la classe de
médicaments
Les événements thromboemboliques artériels (ETA) sont des
effets indésirables potentiellement liés à l'inhibition systémique du VEGF. Il
existe un risque théorique d'ETA, y compris d'accident vasculaire cérébral et
d'infarctus du myocarde, suite à l'utilisation intravitréenne d'inhibiteurs du
VEGF. L'incidence des ETA était faible au cours des études cliniques avec
l'aflibercept chez les patients atteints de DMLAn et d'OMD. Aucune différence
notable n'a été observée entre les groupes traités par Eylea 114,3 mg/mL et les
groupes comparateurs traités par Eylea 40 mg/mL.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
SURVEILLANCE après injection intravitréenne :
- pression intraoculaire,
- perfusion de la tête du nerf optique.
- pression intraoculaire,
- perfusion de la tête du nerf optique.
PREVENIR
IMMEDIATEMENT le médecin en cas de :
- rougeur de l'œil,
- douleur oculaire,
- sensation de gêne accrue,
- vision floue ou baisse de vision,
- augmentation de la sensibilité à la lumière.
CONTACTER IMMEDIATEMENT le médecin en cas de :
- opacification du cristallin,
- saignement à l'arrière de l'œil,
- augmentation de la pression à l'intérieur de l'œil,
- saignement dans l'œil,
- décollement, déchirure ou saignement de la couche sensible à la lumière située à l'arrière de l'oeil, entraînant des éclairs lumineux avec des corps flottants, pouvant parfois aller jusqu'à une perte de la vision.
FEMME EN AGE DE PROCREER : UTILISER une contraception efficace pendant le traitement et pendant au moins 4 mois après la dernière injection.
NE PAS CONDUIRE ou utiliser de machines jusqu'à récupération d'une fonction visuelle suffisante.
- rougeur de l'œil,
- douleur oculaire,
- sensation de gêne accrue,
- vision floue ou baisse de vision,
- augmentation de la sensibilité à la lumière.
CONTACTER IMMEDIATEMENT le médecin en cas de :
- opacification du cristallin,
- saignement à l'arrière de l'œil,
- augmentation de la pression à l'intérieur de l'œil,
- saignement dans l'œil,
- décollement, déchirure ou saignement de la couche sensible à la lumière située à l'arrière de l'oeil, entraînant des éclairs lumineux avec des corps flottants, pouvant parfois aller jusqu'à une perte de la vision.
FEMME EN AGE DE PROCREER : UTILISER une contraception efficace pendant le traitement et pendant au moins 4 mois après la dernière injection.
NE PAS CONDUIRE ou utiliser de machines jusqu'à récupération d'une fonction visuelle suffisante.
Femmes en âge de procréer
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement et pendant au moins 4 mois après la dernière injection intravitréenne d'Eylea 114,3 mg/mL.
Grossesse
Il existe des données limitées sur l'utilisation de l'aflibercept chez la femme enceinte.
Les études menées chez l'animal ont mis en évidence une toxicité sur les fonctions de reproduction (voir rubrique Données de sécurité préclinique).
Eylea 114,3 mg/mL ne doit pas être utilisé pendant la grossesse à moins que le bénéfice attendu pour la mère ne l'emporte sur le risque potentiel pour le fœtus.
Allaitement
Basé sur des données humaines très limitées, de faibles quantités d'aflibercept peuvent être excrétées dans le lait maternel. L'aflibercept est une protéine de haut poids moléculaire et la quantité de médicament absorbée par le nourrisson devrait être limitée. Les effets de l'aflibercept sur un nouveau-né/nourrisson allaité ne sont pas connus.
Par mesure de précaution, l'allaitement n'est pas recommandé pendant l'utilisation d'Eylea 114,3 mg/mL.
Fertilité
Il n'existe aucune donnée sur la fertilité chez l'être humain. Les résultats des études menées chez l'animal avec une exposition systémique élevée indiquent que l'aflibercept peut altérer la fertilité chez le mâle et la femelle (voir rubrique Données de sécurité préclinique).
Aucune étude d'interaction n'a été réalisée.
Eylea doit être administré uniquement par des médecins qualifiés, expérimentés dans l'administration d'injections intravitréennes.
Posologie
La dose recommandée est de 8 mg d'aflibercept, correspondant à 0,07 mL de solution. La posologie est la même pour la DMLAn et pour l'OMD. La dose de 8 mg nécessite d'utiliser Eylea 114,3 mg/mL.
À l'instauration du traitement, Eylea est injecté une fois par mois pendant 3 mois consécutifs. Ensuite, en fonction du jugement du médecin sur les résultats visuels et/ou anatomiques, l'intervalle entre deux injections peut être étendu jusqu'à quatre mois. Par la suite, l'intervalle entre deux injections peut être étendu à cinq mois, comme lors de l'utilisation d'un protocole « Treat and Extend », tout en maintenant des résultats visuels et/ou anatomiques stables (voir rubrique Propriétés pharmacodynamiques).
En cas de détérioration des paramètres visuels et/ou anatomiques, l'intervalle entre deux injections doit être réduit en conséquence, en fonction du jugement du médecin. Pendant la phase d'entretien, l'intervalle le plus court entre 2 injections est de 2 mois.
La réalisation de plus de trois injections mensuelles consécutives d'une dose de 8 mg d'Eylea n'a pas été étudiée.
La fréquence des visites de suivi dépend de l'état du patient et du jugement du médecin. Les événements nécessitant la suspension du traitement sont présentés à la rubrique Mises en garde spéciales et précautions d'emploi.
Populations particulières
Insuffisance rénale ou hépatique
Aucune étude spécifique chez les patients atteints d'insuffisance rénale ou hépatique n'a été menée.
Les données actuellement disponibles ne suggèrent pas un besoin d'adaptation posologique d'Eylea chez ces patients (voir rubrique Propriétés pharmacocinétiques).
Patients âgés
Les données actuellement disponibles ne suggèrent pas un besoin d'adaptation posologique d'Eylea chez ces patients.
Population pédiatrique
La sécurité et l'efficacité d'Eylea 114,3 mg/mL chez les enfants et les adolescents âgés de moins de 18 ans n'ont pas été établies. L'utilisation d'Eylea 114,3 mg/mL n'est pas justifiée dans la population pédiatrique dans les indications DMLAn et OMD.
Mode d'administration
Eylea doit uniquement être administré par injection intravitréenne.
Les injections intravitréennes doivent être réalisées par des médecins qualifiés et expérimentés dans ce type d'injections, conformément aux bonnes pratiques et aux recommandations en vigueur. De façon générale, il est nécessaire d'assurer une anesthésie et des conditions d'asepsie adéquates, y compris par l'application d'un antibactérien local à large spectre (par ex. povidone iodée sur la zone périoculaire, la paupière et la surface oculaire). La désinfection chirurgicale des mains, le port de gants stériles, l'utilisation d'un champ stérile et d'un spéculum à paupières stérile (ou équivalent) sont recommandés.
L'aiguille pour injection doit être insérée 3,5 à 4,0 mm en arrière du limbe dans la cavité vitréenne, en évitant le méridien horizontal et en visant le centre du globe oculaire. Le volume de 0,07 mL peut alors être injecté. Un point d'injection scléral différent doit être utilisé lors des injections ultérieures.
Immédiatement après l'injection intravitréenne, les patients doivent être surveillés pour détecter une éventuelle augmentation de la pression intraoculaire. Une surveillance appropriée peut comporter par exemple, une surveillance de la perfusion de la tête du nerf optique ou une tonométrie. Si nécessaire, un équipement stérile de paracentèse doit être disponible.
Après l'injection intravitréenne, les patients doivent être informés qu'ils doivent signaler sans délai tout symptôme évocateur d'endophtalmie (par ex. douleur oculaire, rougeur de l'œil, photophobie, vision trouble).
Chaque flacon ou seringue préremplie doit uniquement être utilisé(e) pour le traitement d'un seul œil.
Après injection, éliminer tout produit non utilisé ou déchet conformément à la réglementation locale en vigueur.
Pour la manipulation du médicament avant administration, voir rubrique Précautions particulières d'élimination et de manipulation.
Durée de conservation :
2 ans
Précautions particulières de conservation :
Eylea 114,3 mg/mL, solution injectable en seringue préremplie
À conserver au réfrigérateur (entre 2 °C et 8 °C).
Ne pas congeler.
Conserver la seringue préremplie dans son blister et dans l’emballage extérieur à l’abri de la lumière.
Avant utilisation, le blister non ouvert peut être conservé à l’extérieur du réfrigérateur en dessous de 25 °C pendant 24 heures maximum.
À conserver au réfrigérateur (entre 2 °C et 8 °C).
Ne pas congeler.
Conserver la seringue préremplie dans son blister et dans l’emballage extérieur à l’abri de la lumière.
Avant utilisation, le blister non ouvert peut être conservé à l’extérieur du réfrigérateur en dessous de 25 °C pendant 24 heures maximum.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Un surdosage par injection d'un volume trop important peut entraîner une augmentation de la pression intraoculaire. Par conséquent, en cas de surdosage, la pression intraoculaire doit être surveillée et, si cela est jugé nécessaire par le médecin ayant procédé à l'injection, un traitement adéquat doit être instauré (voir rubriques Mises en garde spéciales et précautions d'emploi et Précautions particulières d’élimination et de manipulation).
Classe pharmacothérapeutique : médicaments ophtalmiques/médicaments contre la néovascularisation, Code ATC : S01LA05.
L'aflibercept est une protéine de fusion recombinante composée des fragments des domaines extracellulaires des récepteurs de type 1 et 2 du VEGF humain fusionnés au fragment Fc de l'IgG1 humaine.
L'aflibercept est produit dans des cellules ovariennes K1 de hamster chinois (CHO) par la technologie de l'ADN recombinant.
Mécanisme d'action
Le facteur de croissance de l'endothélium vasculaire de type A (VEGF-A) et le facteur de croissance placentaire (PlGF) appartiennent à la famille des facteurs angiogéniques de type VEGF qui agissent comme de puissants facteurs mitogènes et chimiotactiques et favorisent la perméabilité vasculaire des cellules endothéliales. Le VEGF agit par l'intermédiaire de deux récepteurs tyrosine kinase, le VEGFR-1 et le VEGFR-2, présents sur la surface des cellules endothéliales. Le PlGF se lie uniquement au VEGFR1, qui est aussi présent sur la surface des leucocytes. L'activation excessive de ces récepteurs par le VEGF-A peut entraîner une néovascularisation pathologique et une perméabilité vasculaire excessive. Le PlGF peut agir indépendamment en activant le VEGFR-1 de façon à promouvoir une réponse inflammatoire au sein de la rétine, et il est connu pour être augmenté dans des affections pathologiques telles que la DMLAn, la rétinopathie diabétique (RD), l'OMD et l'occlusion veineuse rétinienne (OVR).
Effets pharmacodynamiques
L'aflibercept agit comme un leurre soluble de récepteur, qui
se lie au VEGF-A et au PlGF avec une affinité supérieure à celle de leurs
récepteurs naturels et peut ainsi inhiber la liaison et l'activation de ces
récepteurs apparentés.
Dans les études effectuées chez l'animal, l'aflibercept a
permis de prévenir la néovascularisation pathologique et les fuites vasculaires
dans différents modèles de pathologies oculaires.
DMLAn
La DMLAn est caractérisée par une néovascularisation choroïdienne (NVC) pathologique. La fuite de sang et de liquide liée à la NVC peut entraîner un œdème de la rétine et/ou une hémorragie sous/intrarétinienne, provoquant une baisse de l'acuité visuelle.
Les effets pharmacodynamiques de l'aflibercept 114,3 mg/mL administré toutes les 12 (8Q12) et toutes les 16 (8Q16) semaines sont décrits en comparaison avec l'aflibercept 40 mg/mL administré toutes les 8 semaines (2Q8) pour l'indication DMLAn. Ces effets sont représentés par la variation de la taille de la NVC entre l'inclusion et la semaine 12 ; la variation de la surface totale de la lésion entre l'inclusion et les semaines 48, 60 et 96 ; et la variation par rapport à la valeur à l'inclusion de l'épaisseur centrale de la rétine (ECT).
Dans le groupe rassemblant les patients ayant reçu le traitement 8Q12 ou 8Q16, la réduction de la taille de la NVC (moyenne des moindres carrés, d'après un modèle mixte à mesures répétées [MMMR]) à la semaine 12 était de -1,63 mm², contre -1,17 mm² chez les patients ayant reçu le traitement 2Q8.
Tableau 2 : Paramètre pharmacodynamique (ensemble d'analyse intégral) dans l'étude PULSAR
|
Résultats d'efficacité |
Semaine |
Eylea 8Q12 (N = 335) |
Eylea 8Q16 (N = 338) |
Eylea 2Q8 (N = 336) |
|
Variation de la surface totale de la lésion par rapport à l'inclusion [mm2] |
|
|||
|
MMC A |
12 |
- 0,55 |
- 0,30 |
|
|
Moyenne arithmétique (ET), observée |
48 |
-0,4 (2,9) |
-0,2 (3,1) |
0,1 (3,6) |
|
MMC (ES)A |
-0,46 (0,19) |
-0,35 (0,20) |
0,09 (0,22) |
|
|
Différence des MMC (IC à 95 %)A,B |
-0,55 (-1,04 ; -0,06) |
-0,44 (-0,94 ; -0,06) |
|
|
|
Moyenne arithmétique (ET), observée |
60 |
-0,5 (2,8) |
-0,4 (3,2) |
-0,3 (3,2) |
|
MMC (ES)A |
-0,48 (0,20) |
-0,54 (0,21) |
-0,24 (0,20) |
|
|
Différence des MMC (IC à 95 %)A,B |
-0,24 (-0,72 ; 0,24) |
-0,29 (-0,79 ; 0,20) |
|
|
|
Moyenne arithmétique (ET), observée |
96 |
-0,3 (3,3) |
-0,3 (3,2) |
-0,2 (3,4) |
|
MMC (ES)A |
|
-0,43 (0,20) |
-0,42 (0,20) |
-0,18 (0,20) |
|
Différence des MMC (IC à 95 %)A,B |
|
-0,25 (-0,72 ; 0,21) |
-0,24 (-0,71 ; 0,22) |
|
B La différence absolue se calcule comme suit : groupes Eylea 8Q12 ou 8Q16 moins groupes 2Q8, respectivement.
ES : erreur standard
ET : écart type
IC : intervalle de confiance
MMC : moyenne des moindres carrés
Figure 1 : Variation de la moyenne des moindres carrés (MMC) de
l'épaisseur centrale rétinienne (ECR) entre l'inclusion et la semaine 96
(ensemble d'analyse intégral) au cours de l'étude PULSAR
OMD
L'œdème maculaire diabétique se caractérise par une augmentation de la perméabilité vasculaire et par des lésions des capillaires rétiniens, ce qui peut entrainer une baisse de l'acuité visuelle.
Les effets pharmacodynamiques de l'aflibercept 114,3 mg/mL administré toutes les 12 (8Q12) et toutes les 16 (8Q16) semaines sont décrits en comparaison avec l'aflibercept 40 mg/mL administré toutes les 8 semaines (2Q8) pour l'indication OMD. Ces effets sont représentés par la variation de la surface de fuite entre l'inclusion et les semaines 48, 60 et 96.
Tableau 3 : Paramètre pharmacodynamique (ensemble d'analyse intégral) dans l'étude PHOTON
|
Résultats d'efficacité |
Semaine |
Eylea 8Q12 (N = 328) |
Eylea 8Q16 (N = 163) |
Eylea 2Q8 (N = 167) |
|
Variation de la surface de fuiteA par rapport à l'inclusion [mm2] |
|
|
||
|
Moyenne arithmétique (ET), observée |
48 |
-13,9 (13,91) |
-9,4 (11,50) |
-9,2 (12,11) |
|
60 |
-13,9 (13,54) |
-12,0 (13,26) |
-14,4 (12,89) |
|
|
96 |
-12,8 (10,98) |
-9,4 (10,61) |
-11,9 (11,26) |
|
ET : écart type
Figure 2 : Variation de la moyenne des moindres carrés (MMC) de
l'épaisseur centrale rétinienne (ECR) entre l'inclusion et la semaine 96 (ensemble
d'analyse intégral) au cours de l'étude PHOTON
Immunogénécité
Après l'administration d'Eylea 114,3 mg/mL pendant un
traitement allant jusqu'à 96 semaines, des anticorps dirigés contre Eylea 114,3
mg/mL ont été détectés chez 2,5 % à 4,4 % des patients traités pour un OMD et une DMLAn. Aucune preuve de l'impact des anticorps
anti-médicament sur la pharmacocinétique, l'efficacité ou la sécurité n'a été
observée.
Efficacité et sécurité cliniques
DMLAn
Objectifs de l'étude
La sécurité et l'efficacité d'Eylea 114,3 mg/mL ont été
évaluées au cours d'une étude multicentrique, randomisée, en double aveugle,
contrôlée contre comparateur actif (étude PULSAR) chez des patients atteints de
DMLAn naïfs de traitement.
L'objectif principal consistait à déterminer si le
traitement par Eylea 114,3 mg/mL administré toutes les 12 (8Q12) ou 16 semaines
(8Q16) permettait d'obtenir une variation de la meilleure acuité visuelle
corrigée (MAVC) non inférieure à celle obtenue avec Eylea 40 mg/mL administré
toutes les 8 semaines chez des patients présentant une DMLAn.
Les objectifs secondaires consistaient à déterminer
l'effet d'Eylea 114,3 mg/mL par rapport à Eylea 40 mg/mL sur les mesures
anatomiques et autres mesures visuelles de la réponse, et à évaluer la
sécurité, l'immunogénicité et la pharmacocinétique de l'aflibercept.
Le critère principal d'efficacité était la variation de la
MAVC entre l'inclusion et la semaine 48, telle que mesurée par le score sur
l'échelle ETDRS (Early Treatment Diabetic Retinopathy Study, étude du
traitement précoce de la rétinopathie diabétique).
Les principaux critères secondaires étaient la variation de
la MAVC entre l'inclusion et la semaine 60, et la proportion de patients sans
liquide intra-rétinien (LIR) ni liquide sous-rétinien (LSR) dans le souschamp
central à la semaine 16.
Les autres critères secondaires comprenaient, entre autres,
la proportion de patients ayant gagné au moins 15 lettres de MAVC entre
l'inclusion et la semaine 48, la proportion de patients ayant obtenu un score
ETDRS d'au moins 69 (équivalent Snellen d'environ 20/40) à la semaine 48, et la
variation du score total au questionnaire sur la fonction visuelle du National
Eye Institute (NEI-VFQ-25) entre l'inclusion et la semaine 48.
Au total, 1 009 patients ont été traités dans l'étude PULSAR. Les patients ont été
randomisés selon un ratio de 1:1:1 dans l'un des 3 groupes de traitement
parallèles :
1.
Eylea 114,3 mg/mL administré toutes les 12 semaines (8Q12)
2.
Eylea 114,3 mg/mL administré toutes les 16 semaines (8Q16)
3.
Eylea 40 mg/mL administré toutes les 8 semaines (2Q8)
Tous les patients ont reçu 3 injections initiales à la dose
qui leur était assignée à 4 semaines d'intervalles. Le protocole prévoyait une
réduction de l'intervalle de traitement chez les patients des groupes 8Q12 et
8Q16 répondant aux deux critères suivants :
1.
Perte de > 5 lettres de MAVC par rapport à la semaine 12, et
2.
Augmentation de > 25 microns de l'ECR par rapport à la semaine 12, ou
hémorragie fovéale ou néovascularisation fovéale nouvellement apparues.
A la semaine 52, indépendamment des variations d'intervalles
dont ils ont bénéficié la 1ère année, tous les patients des groupes 8Q12 et
8Q16 (population per protocol) pouvaient bénéficier d'une extension de leur
intervalle de traitement (par paliers de 4 semaines) s'ils répondaient aux
critères suivants :
1.
Perte de < 5 lettres de MAVC par rapport à la semaine 12, et
2.
Absence de liquide dans le sous-champ central à la tomographie à
cohérence optique (OCT), et
3.
Absence d'hémorragie fovéale ou de néovascularisation fovéale
nouvellement apparues.
Chez les patients qui ne répondaient pas aux critères de
réduction ou d'allongement de l'intervalle de traitement, celui-ci n'était pas
modifié. L'intervalle minimum entre les injections était de 8 semaines dans
l'ensemble des groupes.
Les patients présentant une atteinte bilatérale étaient
éligibles pour recevoir Eylea 40 mg/mL ou un autre médicament anti-VEGF dans
l'œil non étudié.
Caractéristiques des patients à
l'inclusion
L'âge des patients était compris entre 50 et 96 ans, avec un
âge moyen de 74,5 ans.
Environ 92 % (309/335) et 87 % (295/338) des patients
randomisés dans les groupes 8Q12 et 8Q16, respectivement, étaient âgés de 65
ans ou plus, et environ 51 % (172/335) et 51 % (171/338) étaient âgés de 75 ans
ou plus.
Résultats
Les patients des groupes 8Q12, 8Q16 et 2Q8 traités jusqu'à
la semaine 48 ont reçu un nombre médian (moyen) de 6,0 (6,1), 5,0 (5,2) et 7,0
(6,9) injections, respectivement.
À la semaine 48, dans le groupe 8Q12, 79,4 % des patients
ont été maintenus à des intervalles Q12 tandis que dans le groupe 8Q16, 76,6 %
des patients ont été maintenus à des intervalles Q16.
Les patients des groupes 8Q12, 8Q16 et 2Q8 traités jusqu'à
la semaine 60 ont reçu un nombre médian (moyen) de 7,0 (7,1), 6,0 (6,2) et 9,0
(8,8) injections, respectivement.
À la semaine 60, 43,1 % des patients du groupe 8Q12 ont
bénéficié d'un intervalle de traitement étendu à 16 semaines, et 38,5 % des patients du groupe 8Q16 ont
bénéficié d'un intervalle de traitement étendu à 20 semaines.
Les patients des groupes 8Q12, 8Q16 et 2Q8 traités jusqu'à
la semaine 96 ont reçu un nombre médian (moyen) de 9,0 (9,7), 8,0 (8,2) et 13,0
(12,8) injections, respectivement.
À la semaine 96, dans les groupes 8Q12 et 8Q16 réunis, 71,0
% des patients avaient atteint des intervalles de traitement de ≥ 16 semaines,
46,8 % des patients avaient atteint des intervalles de traitement de ≥ 20
semaines et 27,8 % des patients avaient atteint des intervalles de traitement
de 24 semaines tout en maintenant les résultats visuels et anatomiques.
Les traitements 8Q12 et 8Q16 se sont montrés non inférieurs et cliniquement équivalents au traitement 2Q8 en ce qui concerne le critère principal d'efficacité « variation moyenne de la MAVC à la semaine 48 » et le principal critère d'efficacité secondaire « variation moyenne de la MAVC à la semaine 60 ». L'effet du traitement avec Eylea 114,3 mg/ml sur la variation moyenne de la MAVC s'est maintenu jusqu'à la semaine 96.
Par ailleurs, le traitement par Eylea (groupes 8Q12 et 8Q16 réunis) s'est montré supérieur au traitement 2Q8 en ce qui concerne le principal critère d'efficacité secondaire « proportion de patients sans liquide intra-rétinien [LIR] ni liquide sous-rétinien [LSR] dans le sous-champ central à la semaine 16 » (voir tableau 4).
Tableau 4 : Résultats d'efficacité de l'étude PULSAR
|
Résultats d'efficacité |
Semaine |
Eylea 8Q12 (N = 335) |
Eylea 8Q16 (N = 338) |
Eylea 2Q8 (N = 336) |
|
Variation moyenne de la MAVC mesurée sur l'échelle ETDRS par rapport à l'inclusionD |
||||
|
Moyenne arithmétique (ET), observée |
48 |
6,7 (12,6) |
6,2 (11,7) |
7,6 (12,2) |
|
MMC (ES)A |
6,06 (0,77) |
5,89 (0,72) |
7,03 (0,74) |
|
|
Différence des MMC (IC à 95 %)A,B |
-0,97 (-2,87 ; 0,92) |
-1,14 (-2,97 ; 0,69) |
|
|
|
Valeur de p (test unilatéral de noninfériorité avec une marge de 4 lettres)A,B |
0,0009 |
0,0011 |
|
|
|
Moyenne arithmétique (ET), observée |
60 |
6,6 (13,6) |
6,6 (11,7) |
7,8 (12,6) |
|
MMC (ES)A |
6,37 (0,74) |
6,31 (0,66) |
7,23 (0,68) |
|
|
Différence des MMC (IC à 95 %)A,B |
-0,86 (-2,57 ; 0,84) |
-0,92 (-2,51 ; 0,66) |
|
|
|
Valeur de p (test unilatéral de noninfériorité avec une marge de 4 lettres)A,B |
0,0002 |
< 0,0001 |
|
|
|
Moyenne arithmétique (ET), observée |
96 |
5,9 (14,2) |
5,6 (13,7) |
7,4 (13,8) |
|
MMC (ES)A |
5,59 (0,77) |
5,52 (0,75) |
6,60 (0,73) |
|
|
Différence des MMC (IC à 95 %)A,B |
-1,01 (-2,82 ; 0,80) |
-1,08 (-2,87 ; 0,71) |
|
|
|
Patients sans LIR ni LSR dans le sous-champ centralD |
||||
|
Proportion (DOR) |
16 |
63,3 % |
51,6 % |
|
|
Différence ajustée de la proportion (IC à 95 %)B,C |
11,7 % (5,3 % ; 18,2 %) |
|
||
|
Valeur de p (test unilatéral de supériorité)B, C |
0,0002 |
|
||
|
Proportion (DOR) |
48 |
71,1 % |
66,8 % |
59,4 % |
|
Différence ajustée de la proportion (IC à 95 %)B,C |
11,7 % (4,5 % ; 18,9 %) |
7,5 % (0,1 % ; 14,8 %) |
|
|
|
Proportion (DOR) |
60 |
74,6 % |
72,2 % |
74,6 % |
|
Différence ajustée de la proportion (IC à 95 %)B,C |
0,0 % (-6,6 % ; 6,7 %) |
-2,2 % (-8,9 % ; 4,4 %) |
|
|
|
Proportion (DOR) |
96 |
69,6 % |
63,6 % |
66,5 % |
|
Différence ajustée de la proportion (IC à 95 %)B,C |
3,0 % (-4,1 % ;10,1 %) |
-3,0 % (-10,2 % ;4,2 %) |
|
|
|
Patients ayant obtenu un score ETDRS d'au moins 69 (équivalent Snellen d'environ 20/40)D |
||||
|
Proportion (DOR) |
48 |
56,9 % |
54,3 % |
57,9 % |
|
Différence ajustée de la proportion (IC à 95 %)B,C |
-0,2 % (-6,6 % ; 6,2 %) |
-2,2 % (-8,4 % ; 4,0 %) |
|
|
|
Proportion (DOR) |
60 |
56,3 % |
54,6 % |
58,2 % |
|
Différence ajustée de la proportion (IC à 95 %)B,C |
-1,1 % (-7,5 % ; 5,3 %) |
-2,3 % (-8,7 % ; 4,1 %) |
|
|
|
Proportion (DOR) |
96 |
53,3 % |
53,1 % |
56,7 % |
|
Différence ajustée de la proportion (IC à 95 %)B,C |
-2,7 % (-9,4 % ; 4,0 %) |
-2,4 % (-9,1 % ; 4,2 %) |
|
|
|
Résultats d'efficacité |
Semaine |
Eylea 8Q12 (N = 335) |
Eylea 8Q16 (N = 338) |
Eylea 2Q8 (N = 336) |
|
Patients ayant gagné au moins 15 lettres de MAVC par rapport à l'inclusionD |
|
|||
|
Proportion (DOR) |
48 |
20,7 % |
21,7 % |
22,1 % |
|
Différence ajustée de la proportion (IC à 95 %)B,C |
-1,7 % (-7,8 % ; 4,3 %) |
-0,9 % (-7,0 % ; 5,1 %) |
|
|
|
Proportion (DOR) |
60 |
23,7 % |
23,1 % |
23,3 % |
|
Différence ajustée de la proportion (IC à 95 %)B,C |
0,1 % (-6,2 % ; 6,3 %) |
-0,7 % (-6,9 % ; 5,5 %) |
|
|
|
Proportion (DOR) |
96 |
22,2 % |
22,8 % |
24,2 % |
|
Différence ajustée de la proportion (IC à 95 %)B,C |
-2,4 % (-8,4 % ; 3,6 %) |
-2,0 % (-8,0 % ; 4,1 %) |
|
|
|
Dernier intervalle de traitement assigné |
|
|||
|
Patients à intervalles de traitement ≥Q12 E |
|
|||
|
Proportion (groupes 8Q12 et 8Q16 réunis) |
96 |
87.8% |
n/a |
|
|
Proportion |
86.6% |
89.0% |
n/a |
|
|
Patients à intervalles de traitement ≥Q16 E |
|
|||
|
Proportion (groupes 8Q12 et 8Q16 réunis) |
96 |
71.0% |
n/a |
|
|
Proportion |
63.6% |
78.4% |
n/a |
|
|
Patients à intervalles de traitement ≥Q20 E |
|
|||
|
Proportion (groupes 8Q12 et 8Q16 réunis) |
96 |
46.8% |
n/a |
|
|
Proportion |
40.5% |
53.1% |
n/a |
|
|
Patients à intervalles de traitement Q24 E |
|
|||
|
Proportion (groupes 8Q12 et 8Q16 réunis) |
96 |
27.8% |
n/a |
|
|
Proportion |
24.7% |
30.8% |
n/a |
|
B La différence absolue se calcule comme suit : groupes Eylea 8Q12 ou 8Q16 moins groupes 2Q8, respectivement.
C Différence pondérée entre les traitements calculée par la méthode de Mantel-Haenszel en utilisant les variables de stratification utilisées pour la randomisation (région géographique, MAVC catégorielle à l'inclusion) et IC calculé en utilisant l'approximation normale.
D Ensemble d'analyse intégral
E Ensemble d'analyse de tolérance ; patients considérés comme traités pour la période concernée DOR : dernière observation rapportée
ES : erreur standard
ET : écart type
IC : intervalle de confiance
MMC : moyenne des moindres carrés
Les intervalles de traitement ont été analysés de manières prédéfinie et exploratoire.
Figure 3 : Variation de la moyenne des moindres carrés (MMC) de la MAVC
mesurée sur l'échelle ETDRS entre l'inclusion et la semaine 96 (ensemble
d'analyse intégral) au cours de l'étude PULSAR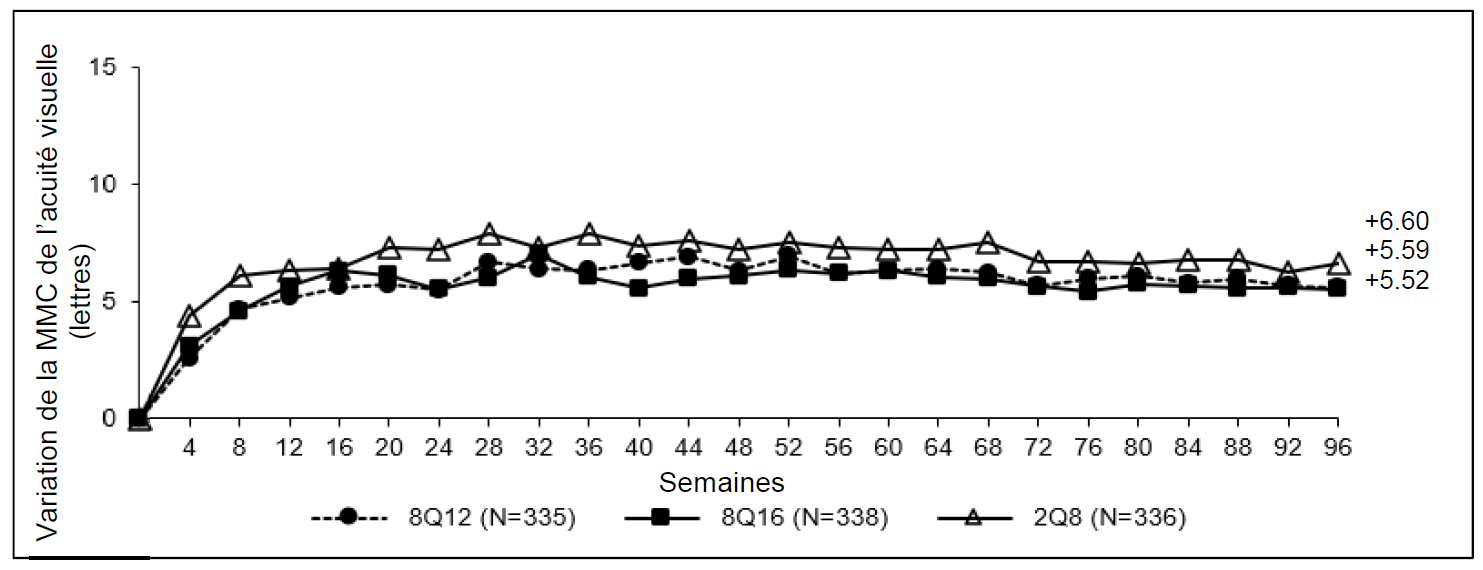
Figure 4 : Dernier intervalle de traitement assigné à la semaine 96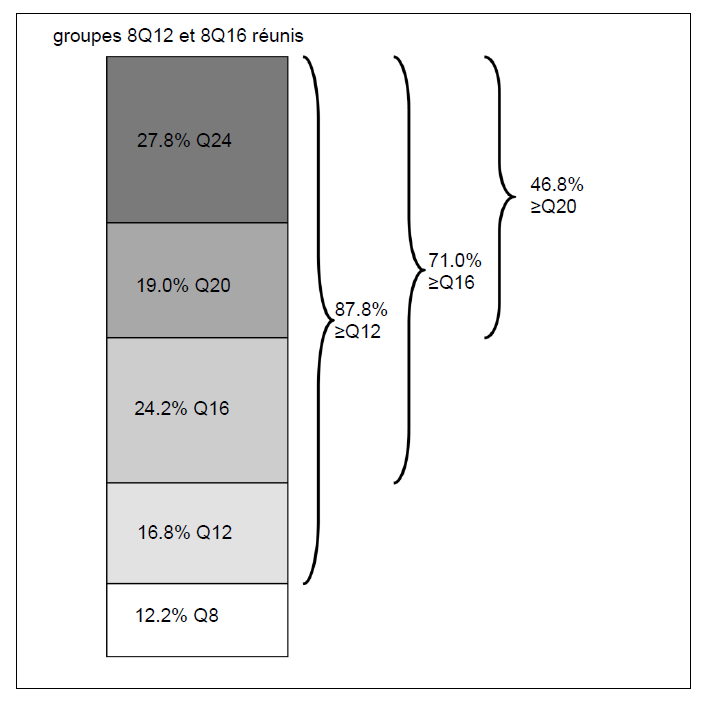
Avec tous les schémas posologiques (8Q12, 8Q16, 2Q8), l'aflibercept a démontré une augmentation significative du score au questionnaire sur la fonction visuelle du National Eye Institute (NEI-VFQ-25) par rapport à l'inclusion, qui était un critère d'efficacité secondaire prédéfini.
Aucune différence cliniquement significative n'a été observée entre les groupes 8Q12, 8Q16 et 2Q8 en termes de variation du score total au questionnaire NEI-VFQ-25 entre l'inclusion et les semaines 48 et 96.
Dans les sous-groupes évaluables basés sur l'âge, le sexe,
la région géographique, l'origine ethnique, l'origine raciale, la MAVC à
l'inclusion et le type de lésion, les résultats d'efficacité étaient cohérents
avec les résultats obtenus dans la population générale.
L'efficacité s'est généralement maintenue jusqu'à la semaine
96.
OMD
Objectifs de l'étude
La sécurité et l'efficacité d'Eylea 114,3 mg/mL ont été
évaluées au cours d'une étude multicentrique, randomisée, en double aveugle,
contrôlée contre comparateur actif (étude PHOTON) chez des patients atteints
d'OMD.
L'objectif principal consistait à déterminer si le
traitement par Eylea 114,3 mg/mL administré toutes les 12 (8Q12) ou 16 semaines
(8Q16) permettait d'obtenir une variation de la MAVC non inférieure à celle
obtenue avec Eylea 40 mg/mL administré toutes les 8 semaines.
Les objectifs secondaires consistaient à déterminer
l'effet d'Eylea 114,3 mg/mL par rapport à Eylea 40 mg/mL sur les mesures
anatomiques et autres mesures visuelles de la réponse, et à évaluer la
sécurité, l'immunogénicité et la pharmacocinétique de l'aflibercept.
Le critère principal d'efficacité était la variation de la
MAVC entre l'inclusion et la semaine 48, telle que mesurée par le score sur
l'échelle ETDRS (Early Treatment Diabetic Retinopathy Study, étude du
traitement précoce de la rétinopathie diabétique).
L'un des principaux critères secondaires était la variation
de la MAVC entre l'inclusion et la semaine 60.
Les autres critères secondaires comprenaient, entre autres,
la proportion de patients ayant gagné au moins 15 lettres de MAVC entre
l'inclusion et la semaine 48, la proportion de patients ayant obtenu un score
ETDRS d'au moins 69 (équivalent Snellen d'environ 20/40) à la semaine 48, et la
variation du score total au questionnaire sur la fonction visuelle du National
Eye Institute (NEI-VFQ-25) entre l'inclusion et la semaine 48.
Au total, 658 patients ont été traités dans l'étude PHOTON.
Les patients ont été randomisés selon un ratio de 2:1 : 1 dans l'un des 3
groupes de traitement parallèles :
1.
Eylea 114,3 mg/mL administré toutes les 12 semaines (8Q12)
2.
Eylea 114,3 mg/mL administré toutes les 16 semaines (8Q16)
3.
Eylea 40 mg/mL administré toutes les 8 semaines (2Q8)
Tous les patients des groupes 8Q12 et 8Q16 ont reçu 3 injections initiales et tous les patients du groupe 2Q8 ont reçu 5 injections initiales à 4 semaines d'intervalles.
Le protocole prévoyait une réduction de l'intervalle de
traitement chez les patients des groupes 8Q12 et 8Q16 répondant aux deux
critères suivants :
1.
Perte de > 10 lettres de MAVC par rapport à la semaine 12 associée à
une persistance ou une aggravation de l'OMD, et
2.
Augmentation de > 50 microns de l'ECR par rapport à la semaine 12.
A la semaine 52, indépendamment des variations d'intervalles
dont ils ont bénéficié la 1ère année, tous les patients des groupes 8Q12 et
8Q16 (population per protocol) pouvaient bénéficier d'une extension de leur
intervalle de traitement (par paliers de 4 semaines) s'ils répondaient aux
critères suivants :
1.
Perte de < 5 lettres de MAVC par rapport à la semaine 12, et
2.
ECR < 300 microns à la SD-OCT (ou < 320 microns si la mesure
englobait l'épithélium pigmentaire rétinien).
Chez les patients qui ne répondaient pas aux critères de
réduction ou d'allongement de l'intervalle de traitement, celui-ci n'était pas
modifié. L'intervalle minimum entre les injections était de 8 semaines dans
l'ensemble des groupes.
Les patients présentant une atteinte bilatérale étaient
éligibles pour recevoir Eylea 40 mg/mL dans l'œil non étudié.
Caractéristiques des patients à
l'inclusion
L'âge des patients était compris entre 24 et 90 ans, avec un
âge moyen de 62,3 ans.
Environ 44 % (143/328) et 44 % (71/163) des patients
randomisés dans les groupes 8Q12 et 8Q16, respectivement, étaient âgés de 65
ans ou plus, et environ 11 % (36/328) et 14 % (14/163) des patients étaient
âgés de 75 ans ou plus.
La proportion de patients ayant déjà été traités pour un OMD
était équilibrée entre les groupes de traitement (43,6 % dans le groupe 8Q12,
43,6 % dans le groupe 8Q16, 44 % dans le groupe 2Q8).
Résultats
Les patients des groupes 8Q12, 8Q16 et 2Q8 traités jusqu'à
la semaine 48 ont reçu un nombre médian (moyen) de 6,0 (6,0), 5,0 (5,0) et 8,0
(7,9) injections, respectivement.
À la semaine 48, dans le groupe 8Q12, 91,0 % des patients
ont été maintenus à des intervalles Q12, tandis que dans le groupe 8Q16, 89,1 %
des patients ont été maintenu à des intervalles Q16.
Les patients des groupes 8Q12, 8Q16 et 2Q8 traités jusqu'à
la semaine 60 ont reçu un nombre médian (moyen) de 7,0 (7,0), 6,0 (6,0) et 10,0
(9,8) injections, respectivement.
À la semaine 60, 42,6 % des patients du groupe 8Q12 ont
bénéficié d'un intervalle de traitement étendu à 16 semaines, et 34,2 % des
patients du groupe 8Q16 ont bénéficié d'un intervalle de traitement étendu à 20
semaines.
Les patients des groupes 8Q12, 8Q16 et 2Q8 traités jusqu'à
la semaine 96 ont reçu un nombre médian (moyen) de 9,0 (9,5), 8,0 (7,8) et 14,0
(13,8) injections, respectivement.
À la semaine 96, dans les groupes 8Q12 et 8Q16 réunis, 72,4
% des patients avaient atteint des intervalles de traitement de ≥ 16 semaines,
44,3 % des patients avaient atteint des intervalles de traitement de ≥ 20
semaines et 26,8 % des patients avaient atteint des intervalles de traitement
de 24 semaines tout en maintenant les résultats visuels et anatomiques.
Le traitement par Eylea (groupes 8Q12 et 8Q16 réunis) s'est montré non inférieur et cliniquement équivalent au traitement 2Q8 en ce qui concerne le critère principal d'efficacité « variation moyenne de la MAVC à la semaine 48 » et le critère principal d'efficacité secondaire « variation moyenne de la MAVC à la semaine 60 ». L'effet du traitement avec Eylea 114,3 mg/ml sur la variation moyenne de la MAVC s'est maintenu jusqu'à la semaine 96.
Tableau 5 : Résultats d'efficacité de l'étude PHOTON
|
Résultats d'efficacité |
Semaine |
Eylea 8Q12 (N = 328) |
Eylea 8Q16 (N = 163) |
Eylea 2Q8 (N = 167) |
|
|
Variation moyenne de la MAVC mesurée sur l'échelle ETDRS par rapport à l'inclusionD |
|
||||
|
Moyenne arithmétique (ET), observée |
48 |
8,77 (8,95) |
7,86 (8,38) |
9,21 (8,99) |
|
|
MMC (ES)A |
8,10 (0,61) |
7,23 (0,71) |
8,67 (0,73) |
||
|
Différence des MMC (IC à 95 %)A,B |
-0,57 (-2,26 ; 1,13) |
-1,44 (-3,27 ; 0,39) |
|
||
|
Valeur de p (test unilatéral de noninfériorité avec une marge de 4 lettres)A,B |
< 0,0001 |
0,0031 |
|
||
|
Moyenne arithmétique (ET), observée |
60 |
9,05 (9,27) |
7,96 (9,14) |
9,62 (9,58) |
|
|
MMC (ES)A |
8,52 (0,63) |
7,64 (0,75) |
9,40 (0,77) |
||
|
Différence des MMC (IC à 95 %)A,B |
-0,88 (-2,67 ; 0,91) |
-1,76 (-3,71 ; 0,19) |
|
||
|
Valeur de p (test unilatéral de noninfériorité avec une marge de 4 lettres)A,B |
0,0003 |
0,0122 |
|
||
|
Moyenne arithmétique (ET), observée |
96 |
8.82 (9.93) |
7.50 (9.86) |
8.41 (11.10) |
|
|
MMC (ES)A |
8.15 (0.63) |
6.59 (0.77) |
7.70 (0.89) |
||
|
Différence des MMC (IC à 95 %)A,B |
-0.45 (-1.55, 2.45) |
-1.11 (-3.27, 1.05) |
|
||
|
Patients ayant obtenu un score ETDRS d'au moins 69 (équivalent Snellen d'environ 20/40)D |
|
||||
|
Proportion (DOR) |
48 |
65,3 % |
62,6 % |
63,0 % |
|
|
Différence ajustée de la proportion (IC à 95 %)B,C |
2,45 % (-6,47 % ; 11,36 %) |
-0,67 % (-11,16 % ; 9,82 %) |
|
||
|
Proportion (DOR) |
60 |
64,7 % |
62,0 % |
60,6 % |
|
|
Différence ajustée de la proportion (IC à 95 %)B,C |
4,34 % (-4,72 % ; 13,40 %) |
1,63 % (-8,91 % ; 12,17 %) |
|
||
|
Proportion (DOR) |
96 |
66.9% |
61.3% |
63.0% |
|
|
Différence ajustée de la proportion (IC à 95 %)B,C |
4.01% (-4.99%, 13.01%) |
-1.51% (-11.91%, 8.89%) |
|
||
|
Patients ayant gagné au moins 15 lettres de MAVC par rapport à l'inclusionD |
|
||||
|
Proportion (DOR) |
48 |
18,7 % |
16,6 % |
23,0 % |
|
|
Différence ajustée de la proportion (IC à 95 %)B,C |
-4,64 % (-12,30 %, 3,02 %) |
-7,14 % (-15,45 %, 1,17 %) |
|
||
|
Proportion (DOR) |
60 |
21,5 % |
16,0 % |
26,1 % |
|
|
Différence ajustée de la proportion (IC à 95 %)B,C |
-5,01 % (-13,04 %, 3,02 %) |
-10,78 % (-19,27 %, -2,29 % ) |
|
||
|
Proportion (DOR) |
96 |
24.5% |
19.6% |
26.1% |
|
|
Différence ajustée de la proportion (IC à 95 %)B,C |
-1.88% (-10.03%, 6.28%) |
-7.07% (-15.94%, 1.80%) |
|
||
|
Résultats d'efficacité |
Semaine |
Eylea 8Q12 (N = 328) |
|
Eylea 8Q16 (N = 163) |
Eylea 2Q8 (N = 167) |
|
Dernier intervalle de traitement assigné |
|
|
|
||
|
Patients à intervalles de traitement ≥Q12 E |
|
|
|
||
|
Proportion (groupes 8Q12 et 8Q16 réunis) |
96 |
|
92.9% |
n/a |
|
|
Proportion |
91.8% |
|
95.0% |
n/a |
|
|
Patients à intervalles de traitement ≥Q16 E |
|
|
|
||
|
Proportion (groupes 8Q12 et 8Q16 réunis) |
96 |
|
72.4% |
n/a |
|
|
Proportion |
64.1% |
|
87.8% |
n/a |
|
|
Patients à intervalles de traitement ≥Q20 E |
|
|
|
||
|
Proportion (groupes 8Q12 et 8Q16 réunis) |
96 |
|
44.3% |
n/a |
|
|
Proportion |
43.0% |
|
46.8% |
n/a |
|
|
Patients à intervalle de traitement Q24 E |
|
|
|
||
|
Proportion (groupes 8Q12 et 8Q16 réunis) |
96 |
|
26.8% |
n/a |
|
|
Proportion |
23.8% |
|
32.4% |
n/a |
|
B La différence absolue se calcule comme suit : groupes Eylea 8Q12 ou 8Q16 moins groupes 2Q8, respectivement.
C Différence pondérée entre les traitements calculée par la méthode de Mantel-Haenszel en utilisant les variables de stratification utilisées pour la randomisation (région géographique, MAVC catégorielle à l'inclusion) et IC calculé en utilisant l'approximation normale.
D Ensemble d'analyse intégral
E Ensemble d'analyse de tolérance ; patients considérés comme traités pour la période concernéeDOR : dernière observation rapportée
ES : erreur standard
ET : écart type
IC : intervalle de confiance
MMC : moyenne des moindres carrés
Les intervalles de traitement ont été analysés de manières prédéfinie et exploratoire.
Figure 5 : Variation de la moyenne des moindres carrés (MMC) de la MAVC
mesurée sur l'échelle ETDRS entre l'inclusion et la semaine 96 (ensemble
d'analyse intégral) au cours de l'étude PHOTON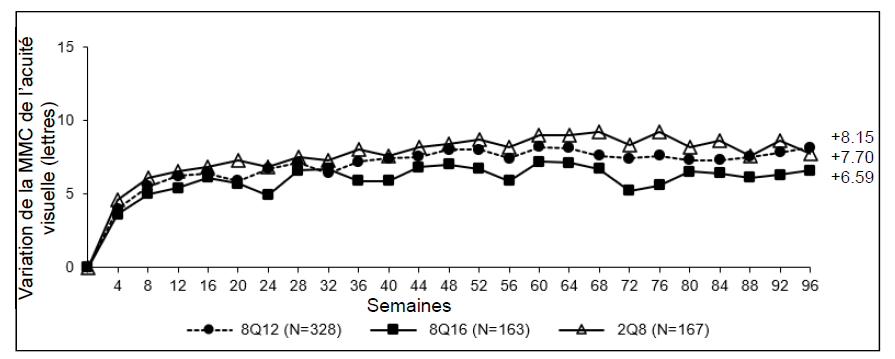
Figure 6 : Dernier intervalle de traitement assigné à la semaine 96
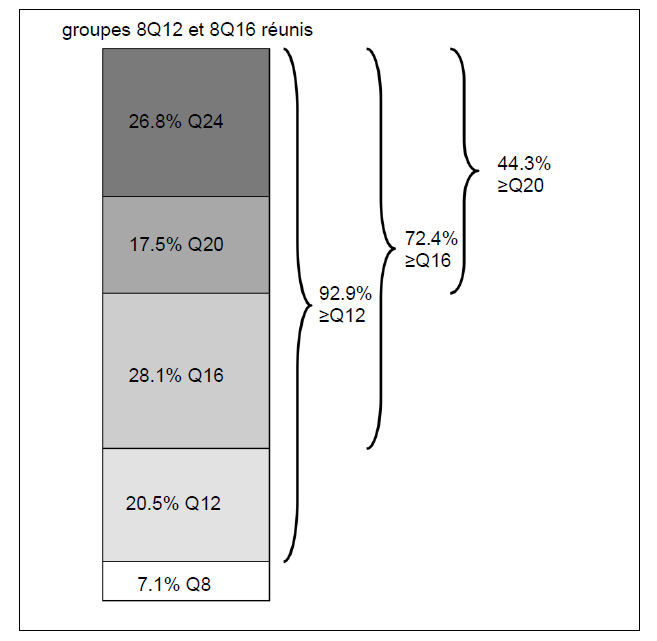
Avec tous les schémas posologiques (8Q12, 8Q16, 2Q8), Eylea
a démontré une augmentation significative du score au questionnaire sur la
fonction visuelle du National Eye Institute (NEI-VFQ-25) par rapport à
l'inclusion, qui était un critère d'efficacité secondaire prédéfini.
Aucune différence cliniquement significative n'a été
observée entre les groupes 8Q12, 8Q16 et 2Q8 en termes de variation du score
total au questionnaire NEI-VFQ-25 entre l'inclusion et les semaines 48 et 96.
Dans les sous-groupes évaluables basés sur l'âge, le sexe, la région géographique, l'origine ethnique, l'origine raciale, la MAVC à l'inclusion, l'ECR à l'inclusion et le traitement antérieur de l'OMD, les résultats d'efficacité étaient cohérents avec les résultats obtenus dans la population générale. L'efficacité s'est généralement maintenue jusqu'à la semaine 96.
Les effets du traitement étaient comparables dans le sous-groupe des patients préalablement traités par rapport aux patients naïfs de traitement.
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec l'aflibercept dans tous les sous-groupes de la population pédiatrique dans la DMLAn et l'OMD (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Absorption / Distribution
L'aflibercept diffuse lentement de l'œil vers la circulation systémique après administration intravitréenne et est essentiellement observé dans la circulation systémique sous forme de complexe inactif stable avec le VEGF ; cependant, seul l'« aflibercept libre » est capable de se lier au VEGF endogène.
Après administration intravitréenne unilatérale de 8 mg d'aflibercept, la Cmax moyenne (ET) de l'aflibercept libre dans le plasma a été de 0,25 (0,21) mg/L et le délai médian d'obtention de la concentration maximale a été de 1 jour, dans les populations atteintes de DMLAn et d'OMD combinées. L'accumulation de l'aflibercept libre dans le plasma après les 3 doses mensuelles initiales a été minime. Aucune accumulation supplémentaire n'a été observée par la suite.
Ces données sont également confortées par des analyses pharmacocinétiques de population.
Élimination
L'aflibercept étant un traitement à base de protéine, aucune étude sur le métabolisme n'a été menée.
L'aflibercept devrait être éliminé à la fois via une élimination médiée par la cible (du fait de sa liaison au VEFG endogène libre) et par métabolisme (via la protéolyse). À la dose de 8 mg administrée par voie intravitréenne, le délai médian d'obtention des dernières concentrations quantifiables d'aflibercept libre dans le plasma était de 3 semaines.
Insuffisance rénale ou hépatique
Aucune étude spécifique chez les patients atteints d'insuffisance rénale ou hépatique n'a été menée avec Eylea 114,3 mg/mL.
L'exposition systémique à l'aflibercept est comparable chez les patients présentant une insuffisance rénale légère à modérée et chez les patients présentant une fonction rénale normale. Les données disponibles chez les patients présentant une insuffisance hépatique légère sont limitées et ne montrent pas d'impact sur l'exposition systémique à l'aflibercept, par rapport aux patients présentant une fonction hépatique normale.
Le traitement par Eylea a une influence mineure sur l'aptitude à conduire des véhicules ou à utiliser des machines du fait de possibles troubles visuels temporaires associés soit à l'injection soit à l'examen de l'œil. Les patients ne doivent pas conduire ou utiliser de machines tant qu'ils n'ont pas récupéré une fonction visuelle suffisante.
Des érosions et des ulcérations de l'épithélium respiratoire dans les cornets nasaux chez les singes traités par aflibercept par voie intravitréenne ont été observées à des expositions systémiques supérieures à l'exposition maximale observée chez l'homme. L'exposition systémique à l'aflibercept libre était environ 46 et 33 fois supérieure, sur la base de la Cmax et de l'ASC, par rapport aux valeurs correspondantes chez les patients adultes après une dose intravitréenne de 8 mg. À la Dose Sans Effet Indésirable Observé (DSEIO) de 0,5 mg/œil chez le singe, l'exposition systémique, basée sur la Cmax et l'ASC, était respectivement 3,2 et 3,8 fois supérieure, par rapport aux valeurs correspondantes chez les patients adultes.
Aucune étude n'a été menée sur le potentiel mutagène ou cancérogène de l'aflibercept.
Un effet de l'aflibercept sur le développement intra-utérin a été mis en évidence dans les études sur le développement embryo-fœtal menées sur des lapines en gestation après administration intraveineuse(3 à 60 mg/kg) et sous-cutanée (0,1 à 1 mg/kg). La DSEIO maternelle était respectivement de 3 mg/kg ou 1 mg/kg. La DSEIO concernant le développement n'a pas été déterminée. À la dose de 0,1 mg/kg, l'exposition systémique à l'aflibercept libre était environ 1,0 et 1,0 fois supérieure, sur la base de la Cmax et de l'ASC cumulée, par rapport aux valeurs correspondantes chez les patients adultes après une injection intravitréenne de 8 mg.
Les effets sur la fertilité chez le mâle et la femelle ont été évalués au cours de l'étude pendant 6 mois chez le singe recevant une administration intraveineuse d'aflibercept à des doses allant de 3 à 30 mg/kg. Une absence ou une irrégularité des menstruations associée à des altérations des niveaux d'hormones reproductives femelles et des modifications dans la morphologie et la mobilité des spermatozoïdes ont été observées à toutes les doses testées. En se basant sur la Cmax et l'ASC pour l'aflibercept libre observées à une dose intraveineuse de 3 mg/kg, les expositions systémiques étaient respectivement environ 377 et 104 fois supérieures par rapport à l'exposition chez l'homme après une dose intravitréenne de 8 mg. Toutes les modifications étaient réversibles.
Eylea 114,3 mg/mL, solution injectable
Le flacon est à usage unique exclusivement pour le traitement d'un seul œil. L'extraction de doses multiples à partir d'un flacon peut augmenter le risque de contamination et d'infection consécutive.
Ne pas
utiliser si l'emballage ou son contenu sont périmés, endommagés ou ont été
altérés.
Contrôler
l'étiquette du flacon pour vérifier qu'il s'agit bien du dosage d'Eylea que vous avez l'intention d'utiliser. La dose de 8 mg
nécessite d'utiliser le flacon d'Eylea 114,3 mg/mL.
L'aiguille
de 18 G à filtre de 5 microns :
- L'aiguille à filtre BD blunt (fill) n'est pas destinée à
l'injection cutanée.
- Ne pas
autoclaver l'aiguille à filtre BD blunt
(fill).
-
L'aiguille à filtre est apyrogène. Ne pas utiliser si l'emballage individuel
est endommagé.
- Jeter
l'aiguille à filtre BD blunt (fill)
usagée dans un collecteur à aiguilles homologué pour objets tranchants.
-
Attention : La réutilisation de l'aiguille à filtre peut entraîner une
infection ou une autre maladie/blessure.
L'injection intravitréenne doit être réalisée à l'aide d'une aiguille
d'injection de 30 G × 13 mm (non fournie).
L'utilisation
d'une aiguille de taille inférieure (de gauge supérieure) à l'aiguille
d'injection de 30 G × 13 mm recommandée pourrait conduire à une augmentation
des forces d'injection.
|
1. |
Avant administration, inspecter la solution injectable. Ne pas utiliser le flacon si des particules sont visibles ou si la solution est trouble ou a changé de couleur. |
|||
|
2. |
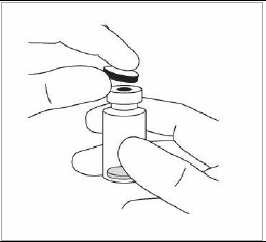
Retirer le capuchon en plastique et désinfecter la partie extérieure du bouchon en caoutchouc du flacon. |
|
||
|
3. |
Utiliser une technique aseptique pour réaliser les étapes 3 à 10. Fixer l'aiguille à filtre, fournie dans l'emballage, sur une seringue Luer Lock stérile de 1 mL. |
|
||
|
4. |
Enfoncer l'aiguille à filtre au centre du bouchon du flacon jusqu'à ce que l'aiguille soit complètement insérée dans le flacon et que son extrémité touche le fond ou les bords du fond du flacon. |
|||
|
5. |
Prélever tout le contenu du flacon d'Eylea dans la seringue, en maintenant le flacon à la verticale et légèrement incliné pour faciliter une complète aspiration. Pour éviter la pénétration d'air, vérifier que le biseau de l'aiguille à filtre est immergé dans le liquide. Continuer à incliner le flacon pendant l'aspiration en gardant le biseau de l'aiguille à filtre immergé dans le liquide. |
|||
|
|
||||
|
6. |
Veiller à tirer suffisamment la tige du piston lors du prélèvement du contenu du flacon de manière à totalement vider l'aiguille à filtre. Après l'injection, tout produit non utilisé doit être éliminé. |
|||
|
7. |
Retirer l'aiguille à filtre et l'éliminer selon la procédure appropriée. Remarque : l'aiguille à filtre ne doit pas être utilisée pour l'injection intravitréenne. |
|||
|
8. |
Fixer fermement l'aiguille pour injection de 30 G × 13 mm sur l'extrémité Luer Lock de la seringue par un mouvement de rotation. |
|
||
|
9 |
En tenant la seringue avec l'aiguille dirigée vers le haut, vérifier l'absence de bulles d'air dans la seringue. Si des bulles sont présentes, tapoter doucement la seringue avec le doigt pour que les bulles remontent jusqu'en haut. |
|
||
|
10 |
Pour
éliminer toutes les bulles et expulser l'excédent de médicament, appuyer
lentement sur le piston de telle manière que le bord plat du piston soit
aligné avec le repère qui indique 0,07 mL
sur la seringue. |
|||


Eylea 114,3 mg/mL, solution injectable en seringue préremplie
La seringue préremplie avec système de dosage OcuClick est à usage unique exclusivement pour le traitement d'un seul œil. L'extraction de plusieurs doses à partir d'une même seringue préremplie avec système de dosage OcuClick pourrait augmenter le risque de contamination et d'infection ultérieure.
Ne pas
utiliser si l'emballage ou son contenu sont périmés, endommagés ou ont été
altérés.
Contrôler l'étiquette de la seringue préremplie avec
système de dosage OcuClick pour vérifier qu'il s'agit
bien du dosage d'Eylea que vous avez l'intention
d'utiliser. La dose de 8 mg nécessite d'utiliser la seringue préremplie d'Eylea 114,3 mg/mL.
L'injection intravitréenne doit être réalisée à l'aide d'une aiguille
d'injection de 30 G × 13 mm (non fournie).
L'utilisation d'une aiguille de taille inférieure (de gauge supérieure) à
l'aiguille d'injection de 30 G × 13 mm recommandée pourrait conduire à une
augmentation des forces d'injection.
|
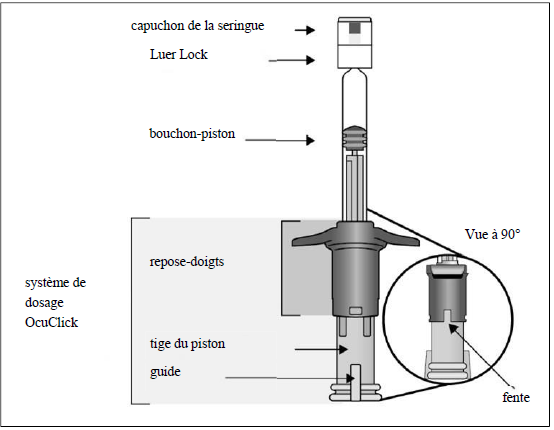
Description de la seringue préremplie avec système de dosage OcuClick intégré
|
||
|
1. |
Préparation Dès que tout est prêt pour l'administration d'Eylea 114,3 mg/mL, ouvrir l'emballage extérieur et en retirer le blister stérile. Ouvrir délicatement le blister en maintenant la stérilité de son contenu. Conserver la seringue sur un plateau stérile jusqu'à la fixation de l'aiguille d'injection. Utiliser une technique aseptique pour réaliser les étapes 2 à 9. |
|
|
2. |
Retrait de la seringue Retirer la seringue du blister stérilisé. |
|
|
3. |
Inspection de la seringue et de la solution injectable Ne pas utiliser la seringue préremplie si - des particules sont visibles ou si la solution est trouble ou a changé de couleur ; - une partie de la seringue préremplie avec système de dosage OcuClick est endommagée ou détachée ; - le capuchon de la seringue s'est détaché de l'adaptateur Luer Lock. |
|
|
4. |
Détachement du capuchon de la seringue Pour détacher le capuchon de la seringue (ne pas le dévisser), tenir la seringue d'une main et le capuchon de la seringue entre le pouce et l'index de l'autre main. Remarque : ne pas tirer sur la tige du piston. |
|
|
5. |
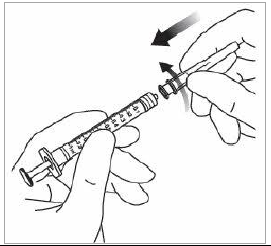
Fixation de l'aiguille Fixer fermement l'aiguille d'injection de 30 G × 13 mm sur l'extrémité Luer Lock de la seringue. |
|
|
6. |
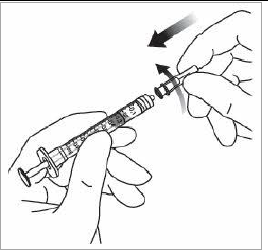
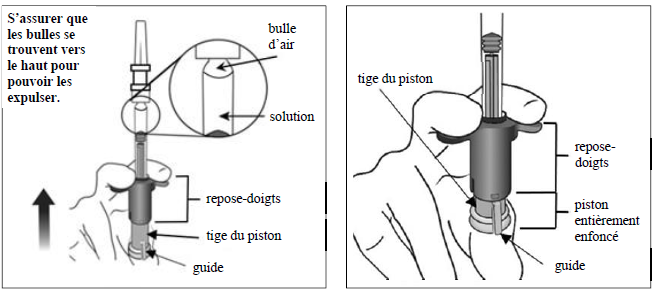
Elimination des bulles d'air En tenant la seringue avec l'aiguille dirigée vers le haut, vérifier l'absence de bulles d'air dans la seringue. Si des bulles sont présentes, tapoter doucement la seringue avec le doigt pour que les bulles remontent jusqu'en haut. |
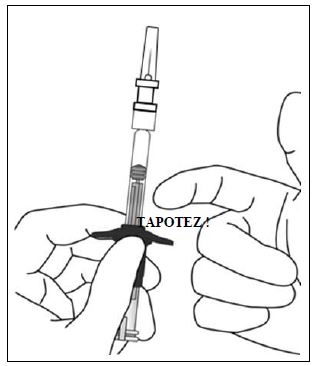 |
|
7. |
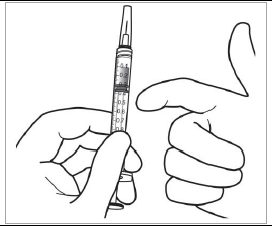
Expulsion de l'air et du volume excédentaire pour amorcer la seringue La seringue ne comporte pas de graduation, car elle est conçue pour régler mécaniquement la dose, comme l'expliquent les étapes ci-dessous. L'amorçage de la seringue et le réglage de la dose doivent être réalisés en suivant les étapes cidessous. Pour éliminer toutes les bulles et expulser l'excédent de médicament, appuyer lentement sur la tige du piston (figure de gauche ci-dessous) jusqu'à ce qu'elle se bloque, c.-à-d. jusqu'à ce que le guide de la tige du piston entre en contact avec le repose-doigts (figure de droite ci-dessous).
|
|
|
8. |
Réglage de la dose Tourner l'extrémité de la tige du piston de 90 degrés dans le sens des aiguilles d'une montre ou dans le sens inverse des aiguilles d'une montre de manière à aligner le guide de la tige du piston au niveau de la fente. Un « clic » peut être entendu. Remarque : le dispositif est maintenant prêt pour l'administration. Ne pas enfoncer la tige du piston avant insertion de l'aiguille dans l'œil. |
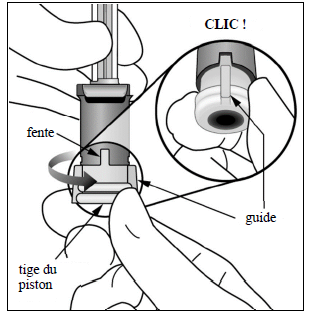 |
|
9. |
Administration et injection Insérer l'aiguille dans le point d'injection oculaire. Injecter la solution en enfonçant la tige du piston jusqu'à ce qu'elle se bloque, c.-à-d. jusqu'à ce que le guide soit entièrement inséré dans la fente. Ne pas appliquer de pression supplémentaire une fois que le guide est inséré dans la fente. Une petite quantité de solution résiduelle peut être visible dans la seringue, ce qui est tout à fait normal. |
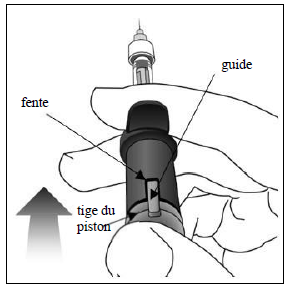 |
|
10. |
La seringue préremplie est prévue pour l'administration d'une seule dose et est à usage unique exclusivement. Après l'injection, éliminer la seringue utilisée dans un collecteur d'aiguilles. |
|


Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste
I
Prescription
réservée aux spécialistes et services OPHTALMOLOGIE
Médicament d'exception.
Remboursement
en fonction de l'indication (JO du 07/02/2025) :
Les seules indications thérapeutiques ouvrant droit à la prise en charge ou au remboursement par l'assurance maladie sont :
- Chez l'adulte, dans le traitement de la forme néovasculaire (humide)
de la dégénérescence maculaire liée à l'âge (DMLA) exsudative
rétrofovéolaire ;
- Chez l'adulte, dans le traitement de la baisse d'acuité visuelle due
à l'œdème maculaire diabétique (OMD), en cas de forme diffuse ou de
fuites proches du centre de la macula, chez les patients dès que
l'acuité visuelle chute à 7/10 Parinaud 3 ou moins et chez lesquels la
prise en charge du diabète a été optimisée.
Solution injectable (injection)
Solution limpide à légèrement opalescente, incolore à jaune pâle, iso-osmotique, de pH 5,8.
Eylea 114,3 mg/mL, solution injectable en seringue préremplie
Seringue préremplie (en verre de type I) munie d’un bouchon-piston gris (caoutchouc élastomère), d’un adaptateur Luer Lock blanc avec un capuchon gris à son extrémité (caoutchouc élastomère) et d’un système de dosage OcuClick bleu (plastique PC/ABS).
Chaque seringue préremplie contient 0,184 mL de solution.
Boîte de 1 seringue préremplie.
Seringue préremplie (en verre de type I) munie d’un bouchon-piston gris (caoutchouc élastomère), d’un adaptateur Luer Lock blanc avec un capuchon gris à son extrémité (caoutchouc élastomère) et d’un système de dosage OcuClick bleu (plastique PC/ABS).
Chaque seringue préremplie contient 0,184 mL de solution.
Boîte de 1 seringue préremplie.
1 mL de solution injectable contient 114,3 mg d'aflibercept*.
Eylea 114,3 mg/mL, solution injectable en seringue préremplie
Chaque seringue préremplie contient 21 mg d'aflibercept dans 0,184 mL de solution. Ceci fournit la quantité nécessaire de produit pour délivrer une seule dose de 0,07 mL contenant 8 mg d'aflibercept.
* L'aflibercept est une protéine de fusion composée des fragments des domaines extracellulaires des récepteurs de type 1 et 2 du VEGF (facteur de croissance de l'endothélium vasculaire) humain fusionnés au fragment Fc de l'IgG1 humaine, produite dans des cellules ovariennes K1 de hamster chinois (CHO) par la technique de l'ADN recombinant.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Saccharose
Chlorhydrate d'arginine
Chlorhydrate d'histidine monohydraté
Histidine
Polysorbate 20
Eau pour préparations injectables