
OPZELURA 15 mg-g, crème, boîte de 1 tube laminé de 100 g
Dernière révision : 19/10/2023
Taux de TVA : 2.1%
Prix de vente : 838,39 €
Taux remboursement SS : 65%
Base remboursement SS : 838,39 €
Laboratoire exploitant : INCYTE BIOSCIENCES FRANCE
Source :

Opzelura est indiqué dans le traitement du vitiligo non-segmentaire avec atteinte faciale chez l'adulte et l'adolescent de plus de 12 ans.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Grossesse et allaitement (voir rubrique Fertilité, grossesse et allaitement).
La crème n'est pas destinée à une administration par voie ophtalmique, orale ou intravaginale (voir rubrique Posologie et mode d'administration). En cas d'exposition accidentelle sur les yeux ou les muqueuses, la crème doit être soigneusement essuyée et/ou rincée avec de l'eau.
Cancer de la peau non-mélanome
Des cas de cancer cutané non-mélanome (CCNM), essentiellement des carcinomes basocellulaires, ont été rapportés chez des patients traités par ruxolitinib en application topique. La plupart de ces patients présentaient des facteurs de risque, tels que des antécédents de photothérapie ou de CCNM. Aucun lien de causalité avec le ruxolitinib n'a été établi. Un examen régulier de la peau est recommandé chez tous les patients, particulièrement chez ceux présentant des facteurs de risque de cancer cutané.
Excipients à effet notoire
Propylène glycol
Ce médicament contient 150 mg de propylène glycol (E1520) pour un gramme de crème, ce qui peut provoquer des irritations cutanées.
Alcool cétylique et alcool stéarylique
Ce médicament contient de l'alcool cétylique et de l'alcool stéarylique susceptibles de provoquer des réactions cutanées locales (par ex. dermatite de contact).
Parahydroxybenzoates
Ce médicament contient du parahydroxybenzoate de méthyle (E218) et du parahydroxybenzoate de propyle susceptibles de provoquer des réactions allergiques (éventuellement retardées).
Butylhydroxytoluène
Ce médicament contient du butylhydroxytoluène (E321) susceptible de provoquer des réactions cutanées locales (par ex. dermatite de contact) ou une irritation des yeux et des muqueuses.
Résumé du profil de sécurité
La sécurité a essentiellement été évaluée dans les études pivotales sur une durée allant jusqu'à un an. Dans l'extension de l'étude à long terme (voir rubrique Propriétés pharmacodynamiques), la sécurité jusqu'à 2 ans était cohérente avec le profil rapporté dans les études pivot. L'effet indésirable le plus fréquent est l'acné au site d'application (5,8 %).
Tableau des effets indésirables
Les effets indésirables sont classés par ordre de fréquence, avec le plus fréquent en première position, selon la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 1 : Effets indésirables
| Classe de systèmes d'organes | Fréquence | Effet indésirable |
| Troubles généraux et anomalies au site d'administration | Fréquent | Acné au site d'application |
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
SURVEILLANCE du traitement :
-si les zones traitées présentent moins de 25 % de repigmentation à 52
semaines de traitement, envisager l'interruption du traitement.
-examen régulier de la peau.
FEMMES EN AGE DE PROCREER : conseiller l'utilisation d'une
contraception efficace pendant toute la durée du traitement et pendant
4 semaines après l'arrêt du traitement.
Contraception chez les femmes en âge de procréer
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant toute la durée du traitement et pendant 4 semaines après l'arrêt du traitement.
Grossesse
Il n'existe pas de données ou il existe des données limitées sur l'utilisation de ruxolitinib chez la femme enceinte. Les données portant sur l'absorption systémique du ruxolitinib par voie topique pendant la grossesse sont insuffisantes. Certains facteurs individuels (par ex. barrière cutanée endommagée, utilisation excessive) pourraient contribuer à une exposition systémique augmentée. Les études chez l'animal ont mis en évidence que le ruxolitinib est embryotoxique et fœtotoxique après une administration par voie orale. Aucune tératogénicité n'a été observée chez les rats ou les lapins (voir rubrique Données de sécurité préclinique). Opzelura est contre-indiqué pendant la grossesse (voir rubrique Contre-indications).
Allaitement
Aucune donnée n'est disponible sur la présence de ruxolitinib dans le lait maternel, sur les effets chez l'enfant allaité ou sur les effets sur la production de lait après l'application topique d'Opzelura. Après une administration par voie orale de ruxolitinib à des rates allaitantes, le ruxolitinib et/ou ses métabolites étaient présents dans le lait à une concentration 13 fois supérieure à la concentration plasmatique maternelle. Dans les études réalisées sur des rats juvéniles, l'administration par voie orale de ruxolitinib a entraîné des effets sur la croissance et les mesures osseuses (voir rubrique Données de sécurité préclinique).
Opzelura est contre-indiqué pendant l'allaitement (voir rubrique Contre-indications) et le traitement doit être interrompu environ 4 semaines avant le début de l'allaitement.
Fertilité
Il n'y a pas de données portant sur l'effet du ruxolitinib sur la fertilité humaine. Dans les études chez l'animal, aucun effet sur la fertilité n'a été observé avec le ruxolitinib par voie orale.
Aucune étude d'interaction n'a été réalisée avec le ruxolitinib administré par voie topique.
Le potentiel d'interactions avec le ruxolitinib est jugé faible en raison de l'exposition systémique limitée après l'administration par voie topique.
Sur la base de données in vitro, le ruxolitinib est principalement métabolisé par le cytochrome P450 3A4 (CYP3A4). Le potentiel d'interaction a été évalué pour le ruxolitinib par voie orale, dans le cadre d'études de pharmacologie clinique dédiées qui impliquaient l'administration d'inhibiteurs du CYP3A4 puissants ou modérés, ou d'un puissant inducteur du CYP3A4. L'ASC plasmatique est presque doublée avec la co-administration d'un puissant inhibiteur du CYP3A4, tandis que seule une légère augmentation est observée en cas de co-administration d'un inhibiteur modéré du CYP3A4.
L'utilisation de ruxolitinib crème en association avec d'autres médicaments topiques destinés au traitement du vitiligo n'a pas été évaluée et leur application concomitante sur les mêmes zones cutanées n'est pas recommandée.
Les autres médicaments topiques utilisés pour le traitement d'autres affections sur les mêmes zones cutanées doivent être appliqués au moins 2 heures après l'application de ruxolitinib crème. Cela s'applique également à l'utilisation d'écran solaire ou d'émollients.
Le traitement par Opzelura doit être instauré et supervisé par des médecins expérimentés dans le diagnostic et le traitement du vitiligo non-segmentaire.
Posologie
Adultes
La dose recommandée est une fine couche de crème appliquée deux fois par jour sur les zones dépigmentées de la peau, sur un maximum de 10 % de la surface corporelle avec un intervalle minimum de 8 heures entre deux applications de ruxolitinib crème. 10 % de la surface corporelle représentent une surface correspondant à 10 fois la paume d'une main avec les 5 doigts. Ruxolitinib crème doit être utilisée sur la plus petite zone de peau nécessaire.
Il convient de ne pas utiliser plus de deux tubes de 100 grammes par mois.
L'obtention d'une repigmentation satisfaisante peut nécessiter un traitement de plus de 24 semaines. Si les zones traitées présentent moins de 25 % de repigmentation à 52 semaines de traitement, l'interruption du traitement devrait être envisagée.
Après obtention d'une repigmentation satisfaisante, le traitement sur ces zones peut être interrompu. Si la dépigmentation revient après l'arrêt du traitement, ce dernier peut être réinstauré sur les zones concernées.
Il n'est pas nécessaire d'arrêter le traitement progressivement.
Populations particulières
Insuffisance hépatique
Aucune étude avec ruxolitinib crème n'a été menée auprès de patients insuffisants hépatiques. Néanmoins, compte tenu de l'exposition systémique limitée, aucune adaptation de dose n'est nécessaire chez les patients insuffisants hépatiques.
Insuffisance rénale
Aucune étude avec ruxolitinib crème n'a été menée auprès de patients insuffisants rénaux. Néanmoins, compte tenu de l'exposition systémique limitée, aucune adaptation de dose n'est nécessaire chez les patients insuffisants rénaux. Par mesure de précaution, ruxolitinib crème ne doit pas être utilisée par des patients atteints d'une insuffisance rénale terminale, en raison du manque de données de sécurité.
Personnes âgées
Un nombre limité de patients âgés de 65 ans et plus ont été inclus dans les études cliniques avec Opzelura dans le vitiligo afin de déterminer s'ils répondaient différemment des sujets plus jeunes (voir rubrique Propriétés pharmacodynamiques). Aucune adaptation de dose n'est requise chez les patients âgés de 65 ans et plus.
Population pédiatrique
Chez les adolescents (12-17 ans), la posologie est la même que chez l'adulte.
La sécurité et l'efficacité de ruxolitinib crème chez les enfants de moins de 12 ans n'ont pas été établies. Aucune donnée n'est disponible.
Mode d'administration
La crème est réservée à une administration par voie cutanée.
Il est préférable de ne pas laver la peau traitée pendant au moins 2 heures après l'application de ruxolitinib crème.
La crème ne doit pas être appliquée sur les lèvres pour éviter tout risque d'ingestion.
Les patients doivent se laver les mains après avoir appliqué la crème, sauf si leurs mains font l'objet du traitement. Si une autre personne applique la crème au patient, elle doit se laver les mains après l'application.
Durée de conservation :
21 mois
Après première ouverture : 6 mois.
Précautions particulières de conservation :
À conserver à une température ne dépassant pas 30 °C.
Sans objet.
Un surdosage après une administration cutanée est peu probable. Si la quantité de crème appliquée est excessive, l'excès peut être essuyé.
En cas d'exposition ophtalmique, buccale ou intravaginale accidentelle, la crème doit être soigneusement essuyée et/ou rincée à l'eau (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Classe pharmacothérapeutique : Autres préparations dermatologiques, agents de la dermatite sauf corticoïdes, Code ATC : D11AH09
Mécanisme d'action
Le ruxolitinib est un inhibiteur de Janus Kinases (JAK) avec une sélectivité pour les isoformes JAK1 et JAK2. La signalisation JAK intracellulaire implique le recrutement de protéines STAT (transducteurs de signal et activateurs de transcription) au niveau des récepteurs de cytokines, et la modulation subséquente de l'expression génique. Les lymphocytes T cytotoxiques auto-immuns producteurs d'IFN? seraient directement responsables de la destruction des mélanocytes dans le vitiligo humain. Le recrutement des lymphocytes cytotoxiques au niveau de la peau lésée est induit par des chimiokines dépendant de l'IFN?, telles que CXCL10. La signalisation en aval de l'IFN? est dépendante de JAK1/2 et le traitement par ruxolitinib réduit les niveaux de CXCL10 chez les patients atteints d'un vitiligo.
Efficacité et sécurité cliniques
Deux études de conception identique, randomisées, en double aveugle, contrôlées contre véhicule (TRuE-V1 et TRuE-V2) ont inclus un total de 674 patients atteints de vitiligo impliquant le visage pour une surface atteinte (faciale et non faciale) n'excédant pas 10 % de la surface corporelle.
L'étendue dépigmentée était comprise entre 3,2 % et 10,1 % de la surface corporelle à l'initiation. Les patients étaient âgés de 12 ans et plus (10,7 % des patients étaient âgés de 12 à 17 ans et 6,7 % étaient âgés de 65 ans ou plus). Les femmes représentaient 53,1 % des patients, 81,9 % des patients étaient Blancs, 4,7 % étaient Noirs et 4,2 % étaient d'origine asiatique. La majorité des patients avaient des peaux de phototype III, IV, V ou VI (67,5 %) selon la classification de Fitzpatrick.
Dans les deux études, les patients, avec une surface atteinte n'excédant pas 10 %, ont été randomisés 2:1 pour recevoir le traitement par ruxolitinib crème ou le véhicule, à raison de deux fois par jour pendant 24 semaines. Cette première phase est suivie d'une phase de traitement pendant 28 semaines supplémentaires, avec ruxolitinib crème à raison de deux fois par jour chez tous les patients. Le critère d'évaluation principal était la proportion de patients obtenant une repigmentation de 75 % évaluée par le Vitiligo Area Scoring Index facial (F-VASI75) en semaine 24. Les critères d'évaluation secondaires clés incluaient les proportions de patients obtenant une repigmentation de 90 % (F-VASI90), une amélioration de 50 % du score Vitiligo Area Scoring Index corporel total (T-VASI50) et un score de 4 ou 5 sur la Vitiligo Noticeability Scale (VNS) (vitiligo « beaucoup moins visible » ou « plus du tout visible »).
La repigmentation des lésions de vitiligo traitées et la supériorité du ruxolitinib crème par rapport au véhicule ont été observées dans les deux études, comme indiquent les différences statistiquement significatives des taux de réponse pour F-VASI75/90, T-VASI50, et le score VNS de 4 ou 5 en semaine 24 (Tableau 2).
La différence d'effet thérapeutique par rapport au véhicule apparaît numériquement dès la semaine 12.
Une repigmentation continue, évaluée par les scores VASI et VNS, a été observée jusqu'en semaine 52 pour les patients ayant continuellement appliqué ruxolitinib crème deux fois par jour depuis le début de l'étude. La proportion de patients ayant obtenu un score F-VASI75 sur la période de traitement de 52 semaines dans les données poolées provenant des études TRuE-V1 et TRuE-V2 est présentée dans la Figure 1.
Des réponses thérapeutiques similaires sont observées en semaine 52 chez ceux qui sont passés du véhicule au ruxolitinib (Figure 1).
Tableau 2 : Pourcentage de patients atteints de vitiligo ayant rempli les critères d'évaluation principal et secondaires clés à 24 semaines (intention de traiter)a
| TRuE-V1 | TRuE-V2 | |||
| Opzelura | Véhicule | Opzelura | Véhicule | |
| (N = 221) | (N = 109) | (N = 222) | (N = 109) | |
| F-VASI75 (%) | 29,8 | 7,4 | 30,9 | 11,4 |
| Différence de taux de réponse (IC à 95 %) | 22,3b (14,214, 30,471) | - | 19,5c (10,537, 28,420) | - |
| F-VASI90 (%) | 15,3 | 2,2 | 16,3 | 1,3 |
| Différence de taux de réponse (IC à 95 %) | 13,2d (7,497, 18,839) | - | 15,0e (9,250, 20,702) | - |
| T-VASI50 (%) | 20,6 | 5,1 | 23,9 | 6,8 |
| Différence de taux de réponse (IC à 95 %) | 15,5d (8,339, 22,592) | - | 17,1c (9,538, 24,721) | - |
| VNS 4 ou 5 (%) | 24,5 | 3,3 | 20,5 | 4,9 |
| Différence de taux de réponse (IC à 95 %) | 21,2c (14,271, 28,143) | - | 15,5d (8,515, 22,561) | - |
a Les critères d'évaluation primaire et secondaires clés ont été corrigés selon la méthode d'imputation multiple.
b p-value < 0,0001
c p-value < 0,001
d p-value < 0,005
e p-value < 0,01
Figure 1 : Proportion de patients obtenant un score F-VASI75 au cours de la période de traitement de 52 semaines (intention de traiter) - données poolées des études TRuE-V1 et TRuE-V2
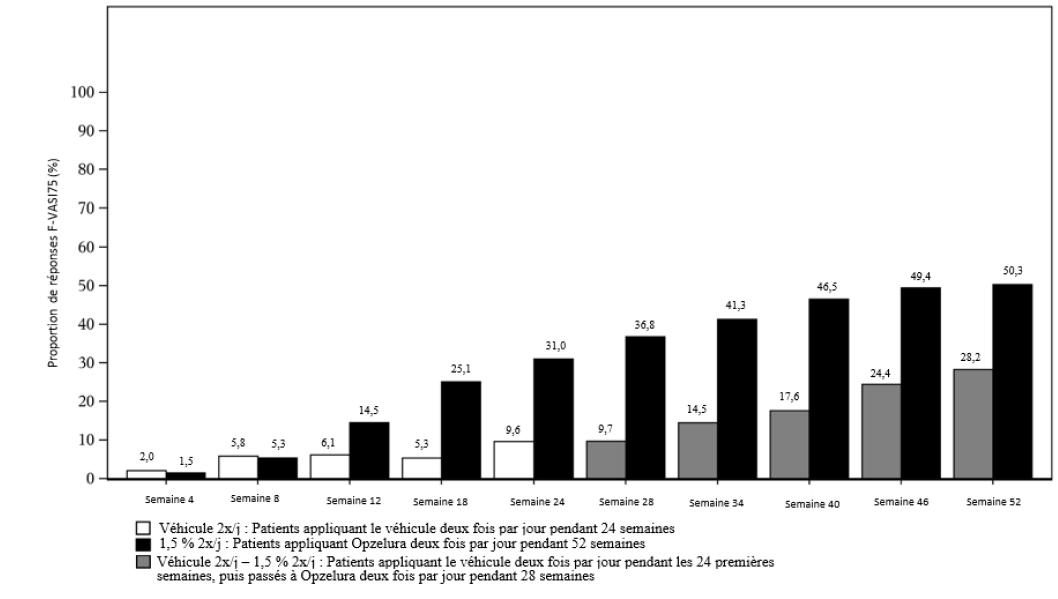
En semaine 52, le taux de réponse observé pour les scores F-VASI90, T-VASI50 et VNS était de 30,3 %, 51,1 %, et 36,3 % respectivement pour la population poolée en ITT.
Durabilité de la réponse
Une étude de phase 3 randomisée, en double aveugle, contrôlée contre véhicule, évaluant l'arrêt et la poursuite du traitement par ruxolitinib crème deux fois par jour incluait 458 patients éligibles présentant un vitiligo et ayant complété l'une des études parentes utilisant le ruxolitinib (TRuE-V1 et TRuE-V1 ; semaine 52) ; les patients ont été affectés dans la cohorte A ou B, avec un suivi allant jusqu'à 104 semaines.
La cohorte A comprenait 116 patients ayant obtenu un ≥ F-VASI90 à la semaine 52 de l'étude parente. Ces patients ont été randomisés à nouveau pour recevoir le ruxolitinib ou le véhicule (c.-à-d.arrêt) jusqu'à apparition d'une rechute dans l'étude (< F-VASI75). Une rechute a été observée chez 15 % des patients du groupe ruxolitinib et 29 % des patients du groupe véhicule. Dans ce groupe, la plupart des rechutes (9/16) sont apparues dans les 4 mois suivant l'arrêt de ruxolitinib crème. Parmi les 16 patients du groupe véhicule qui présentaient une rechute et ont été à nouveau traités, la reprise du traitement a entraîné le retour d'un F-VASI75 chez 12 patients (75 %) en une médiane de 12 semaines et d'un F-VASI90 chez 11 patients (69 %) en une médiane de 15 semaines.
La cohorte B comprenait 342 patients ayant obtenu un < F-VASI90 à la semaine 52 de l'étude parente. Ces patients ont poursuivi le traitement par ruxolitinib en ouvert ; à la semaine 104, parmi les patients initialement randomisés pour recevoir ruxolitinib crème deux fois par jour, 66 % ont obtenu un F- VASI75 et 34 % ont obtenu un F-VASI90.
Population pédiatrique
Un total de 72 adolescents (12 à < 18 ans ; n = 55 ruxolitinib crème, n = 17 véhicule) a été inclus dans les études pivots. Les adolescents traités par ruxolitinib ont présenté des taux de réponse équivalents aux adultes âgés de 18-65 ans.pour les critères d'évaluation principal et secondaires clés à 24 semaines.
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats des études réalisées avec Opzelura dans un ou plusieurs sous-groupes de la population pédiatrique pour le traitement du vitiligo (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Absorption
La pharmacocinétique de ruxolitinib crème a été évaluée chez 429 sujets atteints de vitiligo âgés de 12 ans et plus (12,6 % étaient âgés de 12-17 ans), avec une atteinte moyenne ± ET de la surface corporelle de 7,31 ± 2,02 % (plage de 3,2 % à 10,0 %). Les sujets ont appliqué environ 1,58 mg/cm2 de ruxolitinib crème (l'intervalle de dose était d'environ 0,18 gramme à 8,4 grammes de ruxolitinib crème par application) sur les mêmes zones cutanées deux fois par jour pendant 24 semaines.
Les concentrations plasmatiques minimales moyennes ± ET à l'état d'équilibre étaient de 56,9 ± 62,6 nM avec une projection de l'ASC0-12h à 683 ± 751 h*nM, ce qui correspond à approximativement 25 % de l'ASC0-12h moyenne à l'état d'équilibre (2716 h*nM) après l'administration par voie orale de 15 mg deux fois par jour chez des participants en bonne santé. La biodisponibilité topique moyenne (moyenne géométrique) pour ruxolitinib crème chez les participants atteints de vitiligo dans les données poolées des deux études de Phase 3 était de 9,72 % (5,78 %).
Distribution
Sur la base d'une étude in vitro, le ruxolitinib est lié à 97 % aux protéines plasmatiques humaines, principalement l'albumine.
Biotransformation
Le ruxolitinib est métabolisé par le CYP3A4 et, dans une moindre mesure, par le CYP2C9.
Élimination
La demi-vie d'élimination moyenne du ruxolitinib administré par voie orale est d'environ 3 heures. La demi-vie terminale apparente moyenne du ruxolitinib après l'application topique d'Opzelura a été estimée chez 9 patients adultes et adolescents avec une dermatite atopique présentant une atteinte ≥ 25 % de la surface corporelle. Elle est d'environ 116 heures, ce qui reflète la lenteur d'absorption du médicament plutôt que sa vitesse d'élimination.
Populations particulières
Insuffisance rénale
L'ASC estimée, ajustée selon l'activité pharmacologique du ruxolitinib et de ses métabolites, augmente approximativement d'un facteur deux en cas d'insuffisance rénale terminale (IRT). Par mesure de précaution, compte tenu des données de sécurité insuffisantes en la matière, Opzelura ne doit pas être utilisé par les patients en IRT.
Insuffisance hépatique
Si l'ASC était augmentée après l'administration par voie orale du ruxolitinib aux patients insuffisants hépatiques, il n'y avait pas de lien de causalité clair entre la sévérité de l'insuffisance hépatique et l'augmentation de l'ASC. Aucun ajustement posologique n'est nécessaire pour les patients en insuffisance hépatique.
Ruxolitinib crème n'a aucun effet ou qu'un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Le ruxolitinib a été évalué dans des études de pharmacologie de sécurité, de toxicité à doses répétées, de génotoxicité, de toxicité de la reproduction, et de carcinogénicité après administration par voie orale. D'autres études complémentaires ont été menées après administration dermique sur des miniporcs et des souris. Les organes cibles associés à l'activité pharmacologique du ruxolitinib dans les études de toxicité à doses répétées par voie orale incluent la moelle osseuse, le sang périphérique et les tissus lymphoïdes. Des infections généralement associées à une immunosuppression ont été observées chez le chien. Les marges (basées sur l'ASC non liée) à des niveaux non nocifs dans les études de toxicité chronique étaient environ 6 à 200 fois supérieures chez les rats mâles et femelles, et 10 fois supérieures chez les chiens, par rapport à l'exposition systémique observée chez les patients atteints de vitiligo ayant appliqué 1,5 % de ruxolitinib crème deux fois par jour. Des diminutions délétères de la pression artérielle accompagnées d'augmentations de la fréquence cardiaque ont été observées dans une étude de télémétrie chez le chien, et une diminution délétère du volume par minute a été constatée dans une étude de la fonction respiratoire chez le rat. Les marges (basées sur la Cmax du médicament non lié) à des niveaux non nocifs dans les études sur les chiens et les rats étaient respectivement environ 300 fois et 100 fois supérieures, à l'exposition systémique observée chez les patients atteints de vitiligo ayant appliqué 1,5 % de ruxolitinib crème deux fois par jour. Aucun effet indésirable n'a été observé dans l'évaluation des effets neuropharmacologiques du ruxolitinib chez le rat.
Une étude à doses répétées de 3 mois par voie dermique a révélé une diminution de la numération lymphocytaire chez la souris. Les marges (basées sur l'ASC non liée) à des niveaux non nocifs étaient environ 10 fois supérieures chez les souris mâles et 24 fois supérieures chez les souris femelles, par rapport à l'exposition systémique observée chez les patients atteints de vitiligo ayant appliqué 1,5 % de ruxolitinib crème deux fois par jour. Une diminution non nocive de la numération lymphocytaire périphérique a également été observée chez les miniporcs dans une étude de toxicité de 9 mois par voie dermique. Les marges (basées sur l'ASC non liée) à des niveaux non nocifs chez les miniporcs étaient environ 3 fois supérieures à l'exposition systémique observée chez les patients atteints de vitiligo ayant appliqué 1,5 % de ruxolitinib crème deux fois par jour. Cet effet n'a pas été observé dans une étude de toxicité dermique d'une durée de 3 mois chez des miniporcs. Aucun signe de toxicité systémique n'a été observé chez les miniporcs de Gottingen après l'administration topique de 1,5 % de ruxolitinib crème deux fois par jour pendant une durée allant jusqu'à 9 mois.
Dans les études réalisées sur des rats juvéniles, l'administration par voie orale du ruxolitinib a entraîné des effets sur la croissance et sur les mesures osseuses. Une diminution de la croissance osseuse a été observée à des doses ≥ 5 mg/kg/jour lorsque le traitement était instauré au 7ème jour après la naissance (comparable à celui du nouveau-né humain) et à des doses ≥ 15 mg/kg/jour lorsque le traitement était instauré au 14ème ou au 21ème jour après la naissance (comparable à celui de l'enfant humain en bas- âge, de 1 à 3 ans). Des fractures et un sacrifice précoce chez le rat ont été observés à des doses ≥ 30 mg/kg/jour lorsque le traitement était instauré au 7ème jour après la naissance. D'après l'ASC non liée, l'exposition à la NOAEL (dose sans effet indésirable observé) chez les rats juvéniles traités dès le 7ème jour après la naissance était environ 20 fois supérieure à celle des patients adultes atteints de vitiligo, tandis qu'une croissance osseuse diminuée et des fractures survenaient respectivement à des expositions 22 à 150 fois supérieures à celle des patients adultes atteints de vitiligo. Les effets étaient généralement plus graves chez les mâles et lorsque l'administration avait débuté plus tôt dans la période post-natale. Hormis le développement osseux, les effets du ruxolitinib chez les rats juvéniles étaient similaires à ceux observés chez les rats adultes. Les rats juvéniles sont plus sensibles que les rats adultes à la toxicité du ruxolitinib.
Dans les études sur le développement embryo-fœtal, l'administration par voie orale de ruxolitinib à des rats et des lapins pendant la gestation a entraîné une diminution du poids du fœtus et une augmentation des pertes post-implantation aux doses associées à une toxicité maternelle. Aucun effet tératogène n'a été mis en évidence chez le rat et le lapin. Les marges (basées sur l'ASC non liée) à des niveaux non nocifs de toxicité sur le développement étaient environ 25 fois supérieures chez les rats, par rapport à l'exposition systémique observée chez les patients atteints de vitiligo ayant appliqué 1,5 % de ruxolitinib crème deux fois par jour. Aucun effet sur la fertilité des rats mâles et femelles n'a été mis en évidence avec le ruxolitinib par voie orale. Dans une étude du développement pré- et post- natal, un léger allongement de la période de gestation, une réduction du nombre de sites d'implantation et une réduction du nombre de petits mis bas ont été observés. Chez les petits, un poids corporel initial moyen plus faible et une courte période de diminution du gain pondéral moyen ont été observés. Chez les rates allaitantes, le ruxolitinib et/ou ses métabolites ont été excrétés dans le lait à une concentration 13 fois supérieure à la concentration plasmatique maternelle. Le ruxolitinib n'a pas été mutagène ou clastogène. Le ruxolitinib n'a pas montré de potentiel carcinogène après administration par voie topique chez les souris et après administration par voie orale chez les rats Sprague-Dawley et les souris transgéniques Tg.rasH2.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I
Médicament nécessitant une surveillance particulière pendant le traitement
Prescription réservée aux spécialistes et services DERMATOLOGIE
Crème
Crème blanche à blanc cassé.
Tube laminé avec revêtement intérieur en polyéthylène basse densité et haute densité fermé par un bouchon en polypropylène.
Tube de 100 g. Un tube par boîte.
Un gramme de crème contient 15 mg de ruxolitinib (sous forme de phosphate).
Excipients à effet notoire
Propylène glycol (E1520), 150 mg/g de crème
Alcool cétylique, 30 mg/g de crème
Alcool stéarylique, 17,5 mg/g de crème
Parahydroxybenzoate de méthyle (E218), 1 mg/g de crème
Parahydroxybenzoate de propyle, 0,5 mg/g de crème
Butylhydroxytoluène (antioxydant dans de la paraffine molle blanche) (E321)
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Butylhydroxytoluène (antioxydant dans de la paraffine molle blanche) (E321)
Alcool cétylique
Diméthicone (E900)
EDTA calcio-disodique (E385)
Stéarate de glycérol auto-émulsifiant
Macrogol
Triglycérides à chaîne moyenne
Parahydroxybenzoate de méthyle (E218)
Paraffine (E905), Liquide légère
Paraffine (E905), molle blanche
Phénoxyéthanol
Polysorbate 20 (E432)
Propylène glycol (E1520)
Parahydroxybenzoate de propyle
Eau purifiée
Alcool stéarylique
Gomme xanthane (E415)