
TAVNEOS 10 mg, gélule, boîte de 1 flacon de 30
Dernière révision : 15/01/2025
Taux de TVA : 2.1%
Laboratoire exploitant : VIFOR FRANCE
Source :

Tavneos, en association avec un traitement par rituximab ou cyclophosphamide, est indiqué chez les patients adultes atteints de granulomatose avec polyangéite (GPA) ou de polyangéite microscopique (PAM) sévère et active (voir rubrique Posologie et mode d'administration).
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Hépatotoxicité
Des effets indésirables graves d'élévation des transaminases hépatiques avec élévation de la bilirubine totale ont été observés chez des patients recevant de l'avacopan en association avec du cyclophosphamide (suivi d'azathioprine ou de mycophénolate) ou du rituximab, et du triméthoprime et du sulfaméthoxazole. Après commercialisation, des cas d'atteinte hépatique d'origine médicamenteuse et de syndrome des voies biliaires évanescentes (VBDS, vanishing bile duct syndrome), y compris des cas d'issue fatale, ont été rapportés (voir rubrique Effets indésirables).
Les transaminases hépatiques et la bilirubine totale doivent être obtenues avant l'instauration du traitement.
L'avacopan doit être évité chez les patients présentant des signes de maladie hépatique, tels qu'une élévation des ASAT, ALAT, de la phosphatase alcaline (PAL) ou de la bilirubine totale > 3 fois la LSN.
Les transaminases hépatiques et la bilirubine totale des patients doivent être surveillées au moins toutes les 4 semaines après le début du traitement pendant les 6 premiers mois, et, par la suite, si cliniquement indiqué (voir rubrique Posologie et mode d'administration).
Sang et système immunitaire
Une numération des globules blancs doit être effectuée ant l'instauration du traitement et les patients doivent être surveillés si cliniquement indiqué et dans le cadre du suivi de routine de l'état sous-jacent du patient (voir rubrique Posologie et mode d'administration).
Le traitement par l'avacopan ne doit pas être instauré si la numération des globules blancs est < 3,5 × 109/L, ou la numération des neutrophiles est < 1,5 × 109/L, ou la numération des lymphocytes est < 0,5 × 109/L.
Les patients recevant de l'avacopan doivent signaler immédiatement tout signe d'infection, d'ecchymose inattendue, de saignement ou toute autre manifestation d'insuffisance de la moelle osseuse.
Infections graves
Des infections graves ont été rapportées chez des patients recevant des associations de traitements pour prendre en charge la GPA ou la PAM, notamment l'avacopan en association avec le rituximab ou le cyclophosphamide (voir rubrique Effets indésirables).
Les patients doivent être évalués pour toute infection grave.
L'avacopan n'a pas été étudié chez les patients atteints d'hépatite B, d'hépatite C ou infectés par le virus de l'immunodéficience humaine (VIH). Avant et pendant le traitement, les patients doivent indiquer à leur médecin s'ils sont atteints de tuberculose, d'hépatite B, d'hépatite C ou d'une infection par le VIH.
Soyez prudent lors du traitement de patients ayant des antécédents de tuberculose, d'hépatite B, d'hépatite C ou infectés par le VIH.
L'avacopan ne diminue pas la formation du complexe d'attaque membranaire (C5b-9) ou complexe terminal du complément (CTC). Aucun cas de Neisseria meningitidis n'a été identifié au cours du programme clinique de l'avacopan. Surveiller les patients traités pour une vascularite associée aux ANCA selon la pratique standard afin de détecter tout signe et symptôme clinique d'infections à Neisseria.
Prophylaxie de la pneumonie à Pneumocystis jirovecii
La prophylaxie de la pneumonie à Pneumocystis jirovecii est recommandée chez les patients adultes atteints de GPA ou de PAM pendant le traitement par l'avacopan, le cas échéant, conformément aux recommandations locales de pratique clinique.
Immunisation
La sécurité de l'immunisation par des vaccins vivants, après un traitement par l'avacopan, n'a pas été étudiée.
Administrer les vaccins de préférence avant l'instauration du traitement par l'avacopan ou pendant la phase de rémission de la maladie.
Angiœdème
Des cas d'angiœdème ont été rapportés chez des patients recevant de l'avacopan (voir rubrique Effets indésirables).
Les patients doivent contacter leur médecin s'ils présentent des symptômes tels qu'un gonflement du visage, des lèvres ou de la langue, une sensation de gorge serrée ou des difficultés à respirer.
L'avacopan doit être suspendu en cas d'angiœdème.
Interaction avec les inducteurs puissants du CYP3A4
L'utilisation d'inducteurs puissants de l'enzyme CYP3A4 (par exemple, carbamazépine, enzalutamide, mitotane, phénobarbital, phénytoïne, rifampicine et millepertuis) avec l'avacopan est à éviter (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Les patients nécessitant une administration sur une longue durée de ces médicaments ne doivent pas être traités par l'avacopan.
Si la co-administration sur une courte durée ne peut être évitée chez un patient déjà sous avacopan, le patient doit être étroitement surveillé en cas de rechute de la maladie.
Troubles cardiaques
Les patients atteints de GPA ou de PAM sont à risque de troubles cardiaques tels que l'infarctus du myocarde, l'insuffisance cardiaque et la vascularite cardiaque.
Des événements indésirables graves (EIG) de troubles cardiaques ont été rapportés chez des patients traités par avacopan. Un traitement basé sur l'association avec le cyclophosphamide suivi de l'azathioprine peut entraîner un risque accru de troubles cardiaques par rapport à un traitement basé sur l'association avec le rituximab.
Tumeur maligne
Les médicaments immunomodulateurs peuvent augmenter le risque de tumeurs malignes. Les données cliniques sont actuellement limitées (voir rubrique Propriétés pharmacodynamiques).
Teneur en hydroxystéarate de macrogolglycérol
Ce médicament contient de l'hydroxystéarate de macrogolglycérol, qui peut provoquer des maux d'estomac et des diarrhées.
Résumé du profil de sécurité
Les
effets indésirables les plus fréquents sont les nausées (23,5 %), les
céphalées (20,5 %), la diminution du nombre de globules blancs (18,7
%), les infections des voies respiratoires supérieures (14,5 %), les
diarrhées (15,1 %), les vomissements (15,1 %) et la rhinopharyngite
(15,1 %).
Les effets indésirables graves les plus fréquents sont les anomalies de la fonction hépatique (5,4 %) et la pneumonie (4,8 %).
Liste tabulée des effets indésirables
Les
effets indésirables observés dans l'étude pivot de phase III sur la
vascularite associée aux ANCA et après commercialisation chez les
patients traités par l'avacopan sont répertoriés dans le tableau 1 par
classe de systèmes d'organes (SOC) et par fréquence.
Les fréquences sont définies selon la convention suivante : très
fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1
000 à < 1/100) et indéterminé (la fréquence ne peut pas être estimée
à partir des données disponibles). Au sein de chaque groupe de
fréquence, les effets indésirables sont présentés suivant un ordre
décroissant de gravité.
Tableau 1 : Effets indésirables
| Classe de systèmes d'organes | Très fréquent (≥ 1/10) | Fréquent (≥ 1/100, < 1/10) | Peu fréquent (≥ 1/1 000,< 1/100) | Indéterminé |
| Infections et infestations | Infection des voies respiratoires supérieures, Rhinopharyngite | Pneumonie, Rhinite, Infection des voies urinaires, Sinusite, Bronchite, Gastro-entérite, Infection des voies respiratoires inférieures, Cellulite, Herpès zoster, Grippe, Candidose buccale, Herpès buccal, Otite moyenne | ||
| Affections hématologiques et du systèmelymphatique | Neutropénie1 | |||
| Affections du système nerveux | Céphalées | |||
| Affections gastro- intestinales1 | Nausées, Diarrhée, Vomissements | Douleur abdominale haute | ||
| Affections hépatobiliaires | Tests hépatiques augmentés1,2 | Atteinte hépatique d'origine medicamenteuse1, Syndrome des voies biliairesévanescentes1 | ||
| Affections de la peau et du tissu sous-cutané | Angiœdème1 | |||
| Investigations | Diminution du nombre de globules blancs3 | Créatine phosphokinase sanguine augmentée1 |
1 Voir rubrique « Description de certains effets indésirables ».
2 Alanine aminotransférase augmentée, bilirubine sanguine totale augmentée, anomalies des fonctions hépatiques, gamma-glutamyl transférase augmentée, enzyme hépatique augmentée, transaminases augmentées.
3 Inclut la leucopénie.
2 Alanine aminotransférase augmentée, bilirubine sanguine totale augmentée, anomalies des fonctions hépatiques, gamma-glutamyl transférase augmentée, enzyme hépatique augmentée, transaminases augmentées.
3 Inclut la leucopénie.
Description de certains effets indésirables
Hépatotoxicité
Dans l'étude pivot de phase III au cours de laquelle 330 patients ont été traités, 13,3 % des patients du groupe avacopan et 11,6 % des patients du groupe prednisone ont présenté une augmentation du test de la fonction hépatique (TFH).
Dans le groupe avacopan, l'augmentation du TFH a été rapportée dans l'étude de phase III et comprenait des hépatites (1,2 %), des hépatites cholestatiques (0,6 %) dont un patient ayant rapporté à la fois une hépatite et une hépatite cholestatique, des lésions hépatocellulaires (0,6 %) chez un patient présentant une hépatite asymptomatique, des cytolyses et des cholestases anictériques sans insuffisance hépatocellulaire.
Dans l'étude pivot de phase III, les troubles hépatobiliaires étaient plus fréquents chez les patients traités par une association de cyclophosphamide suivie d'azathioprine (10,2 %) par rapport à ceux traités par une association de rituximab (3,7 %).
Le médicament à l'étude a été interrompu ou arrêté définitivement en raison d'une augmentation du TFH chez 5,4 % des patients du groupe avacopan et 3,0 % des patients du groupe prednisone. Des effets indésirables graves liés à une augmentation du TFH ont été rapportés chez 5,4 % des patients du groupe avacopan et 3,7 % des patients du groupe prednisone. Tous les événements hépatiques graves se sont résolus avec le retrait de l'avacopan et/ou d'autres médicaments potentiellement hépatotoxiques, y compris le triméthoprime et le sulfaméthoxazole.
Après commercialisation, des cas d'atteinte hépatique d'origine médicamenteuse et de syndrome des voies biliaires évanescentes (VBDS, vanishing bile duct syndrome) ont été rapportés (voir rubrique Mises en garde spéciales et précautions d'emploi).
Neutropénie
Dans l'étude pivot de phase III, une neutropénie a été rapportée chez 4 patients (2,4 %) dans chaque groupe de traitement.
Un seul cas d'agranulocytose a été rapporté dans le groupe prednisone et dans le groupe avacopan.
Le patient du groupe avacopan présentait une neutropénie centrale sur une biopsie de moelle osseuse qui s'est résolue spontanément sans traitement supplémentaire.
Créatine phosphokinase augmentée
Dans l'étude pivot de phase III, 6 patients (3,6 %) dans le groupe avacopan et 1 patient (0,6 %) dans le groupe prednisone ont présenté une augmentation de la créatine phosphokinase (CPK).
Hypersensibilité incluant un angiœdème
Dans l'étude pivot de phase III, 2 patients (1,2 %) du groupe avacopan ont présenté un angiœdème. Un patient a été hospitalisé pour cet événement. L'avacopan a été interrompu et les deux événements se sont résolus sans séquelles. L'avacopan a été repris chez un patient et l'angiœdème ne s'est pas reproduit.
Troubles gastro-intestinaux
Dans l'étude pivot de phase III, des troubles gastro-intestinaux ont été observés chez 74,6 % des patients traités par l'avacopan en association avec le cyclophosphamide suivi d'azathioprine par rapport à ceux traités en association avec le rituximab (53,3 %).
Populations particulières
Population pédiatrique
Au total, 3 adolescents ont été inclus dans l'étude de phase III, 1 dans le groupe prednisone et 2 dans le groupe avacopan. Il n'existe pas de données chez les enfants âgés de moins de 12 ans (voirrubrique Propriétés pharmacodynamiques).
Patients âgés
Le
profil de sécurité était similaire entre les patients âgés de ≥ 65 ans
et les patients adultes âgés de < 65 ans dans les études cliniques.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
SURVEILLANCE AVANT le traitement :
- Transaminases hépatiques.
- Bilirubine totale.
- Numération des globules blancs.
- Depistage d'une tuberculose, d'hépatite B, d'hépatite C ou d'une infection par le VIH.
PENDANT le traitement :
- Surveiller les transaminases hépatiques et la bilirubine totale au
moins toutes les 4 semaines après le début du traitement pendant les
6 premiers mois, et par la suite, si cliniquement indiqué.
- Surveiller les patients et effectuer une numération
des globules blancs si cliniquement indiqué
et dans le cadre du suivi de routine de la pathologie sous-jacente du
patient.
- La prophylaxie de la pneumonie à Pneumocystis jirovecii est
recommandée, conformément aux recommandations locales de pratique
clinique.
ARRETER LE TRAITEMENT et DEMANDER un avis médical urgent en cas de gonflement du visage, des lèvres, de la langue ou de la gorge ou de difficultés à respirer.
CONSULTER UN MEDECIN en cas de:
- infection, ecchymoses et saignements inattendus,
- sensation de malaise (nausée ou vomissement), sensation de fatigue, perte d'appétit, jaunissement de la peau ou des yeux, urines foncées, démangeaisons ou douleur dans la partie supérieure de l'estomac.
EVITER de consommer du pamplemousse ou du jus de pamplemousse pendant le traitement.
EVITER de consommer des préparations à base de plantes contenant du millepertuis (Hypericum perforatum).
Femmes en âge de procréer : UTILISER une contraception efficace pendant le traitement.
Grossesse
Il n'existe pas de données sur l'utilisation de l'avacopan chez la femme enceinte.
Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique Données de sécurité préclinique).
L'avacopan n'est pas recommandé pendant la grossesse et chez les femmes en âge de procréer n'utilisant pas de contraception.
Allaitement
Chez l'animal, l'avacopan n'a pas été mesuré dans le lait des femelles allaitantes ; cependant, l'avacopan a été détecté dans le plasma de la progéniture des femelles allaitantes sans effets apparents sur la progéniture (voir rubrique Données de sécurité préclinique).
Un risque pour les nouveau-nés/nourrissons ne peut être exclu. Une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre/de s'abstenir du traitement par l'avacopan en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la femme.
Fertilité
Il n'existe aucune donnée sur les effets de l'avacopan sur la fertilité humaine. Les données chez l'animal n'ont pas révélé d'altération de la fertilité chez les mâles ou les femelles (voir rubrique Données de sécurité préclinique).
L'avacopan est un substrat du CYP3A4. La co-administration de l'avacopan avec des inducteurs ou des inhibiteurs de cette enzyme peut affecter la pharmacocinétique de l'avacopan.
Effet des inducteurs puissants du CYP3A4 sur l'avacopan
La co-administration d'avacopan et de rifampicine, un inducteur puissant de l'enzyme CYP3A4, a entraîné une diminution de l'aire sous la courbe (ASC) des concentrations en fonction du temps et de la concentration plasmatique maximale (Cmax) de l'avacopan d'environ 93 % et 79 %, respectivement. Cette interaction pouvant entraîner une perte d'efficacité de l'avacopan, l'utilisation d'inducteurs puissants de l'enzyme CYP3A4 (par exemple, carbamazépine, enzalutamide, mitotane, phénobarbital, phénytoïne, rifampicine et millepertuis) en association à l'avacopan doit être évitée (voir rubrique Mises en garde spéciales et précautions d'emploi). Les patients nécessitant une administration de ces médicaments sur une longue durée ne doivent pas être traités par l'avacopan. Si la co-administration sur une courte durée ne peut être évitée chez un patient déjà sous avacopan, le patient doit être étroitement surveillé pour anticiper toute rechute de la maladie.
Effet des inducteurs modérés du CYP3A4 sur l'avacopan
Faire preuve de prudence lors de l'utilisation d'inducteurs modérés du CYP3A4 (par exemple, le bosentan, l'éfavirenz, l'étravirine et le modafinil) prescrits en association avec l'avacopan et évaluer soigneusement le bénéfice/risque de l'avacopan.
Effet des inhibiteurs puissants du CYP3A4 sur l'avacopan
La co-administration d'avacopan et d'itraconazole, un inhibiteur puissant de l'enzyme CYP3A4, a entraîné une augmentation de l'ASC et de la Cmax de l'avacopan de l'ordre de 2,2 et 1,9 fois, respectivement. Par conséquent, les inhibiteurs enzymatiques puissants du CYP3A4 (par exemple, bocéprévir, clarithromycine, conivaptan, indinavir, itraconazole, kétoconazole, lopinavir/ritonavir, mibéfradil, néfazodone, nelfinavir, posaconazole, ritonavir, saquinavir, télaprévir, télithromycine et voriconazole) doivent être utilisés avec prudence chez les patients traités par avacopan. Les patients doivent être surveillés pour déceler toute augmentation éventuelle des effets indésirables due à l'exposition accrue à l'avacopan.
Le pamplemousse et le jus de pamplemousse peuvent augmenter la concentration de l'avacopan ; par conséquent, le pamplemousse et le jus de pamplemousse doivent être évités chez les patients traités par l'avacopan.
Effet de l'avacopan sur d'autres médicaments
L'avacopan est un inhibiteur modéré du CYP3A4 in vivo et peut augmenter les expositions plasmatiques des médicaments associés qui sont des substrats du CYP3A4 (par exemple, alfentanil, ciclosporine, dihydroergotamine, ergotamine, fentanyl, sirolimus et tacrolimus). Les patients doivent être pris en charge conformément au résumé des caractéristiques du produit des médicaments associés. Des réductions de dose ou une surveillance des événements indésirables peuvent être nécessaires.
Dans une étude clinique, la co-administration d'avacopan et de simvastatine, un substrat sensible duCYP3A4, a entraîné une augmentation de l'ASC et de la C max de la simvastatine de 3,5 et 3,2 fois, respectivement. Veuillez consulter le résumé des caractéristiques du produit de la simvastatine pour les ajustements de dose appropriés.
Effet de l'hydroxystéarate de macrogolglycérol sur les substrats sensibles de la P-glycoprotéine (P-gp).
Un effet cliniquement pertinent de l'excipient hydroxystéarate de macrogolglycérol sur les substrats sensibles de la P-gp ayant une biodisponibilité relativement faible (par exemple, le dabigatran etexilate) ne peut être exclu. Faire preuve de prudence lors de l'utilisation de substrats P-gp à faible biodisponibilité chez les patients traités par avacopan.
Le traitement doit être instauré et surveillé par des professionnels de la santé qualifiés dans le diagnostic et le traitement de la GPA ou de la PAM (voir rubrique Mises en garde spéciales et précautions d'emploi).
Posologie
La dose recommandée de Tavneos est de 30 mg (3 gélules de 10 mg chacune) par voie orale deux fois par jour, matin et soir, avec de la nourriture.
Tavneos doit être administré
en association avec du rituximab ou du cyclophosphamide comme suit :
• rituximab à raison de 4 doses hebdomadaires par
voie intraveineuse ou,
• cyclophosphamide par voie intraveineuse
ou orale pendant 13 ou 14 semaines, suivi d'azathioprine
ou de mycophénolatemofétil
par voie orale et,
• glucocorticoïdes
si cliniquement indiqué.
Pour plus de
détails sur les doses, les traitements concomitants par glucocorticoïdes et les
données sur l'efficacité et la sécurité des associations, voir rubriques Effets
indésirables et Propriétés pharmacodynamiques.
Les données des études cliniques sont limitées à 52 semaines d'exposition suivies de 8 semaines d'observation.
Doses oubliées
Si un patient oublie une dose, celle-ci doit être prise dès que possible, sauf s'il reste moins de trois heures avant la dose suivante prévue. Si le délai est de moins de trois heures avant la dose suivante, la dose oubliée ne doit pas être prise.
Gestion des doses
Le traitement
doit être réévalué cliniquement et arrêté temporairement si :
• les
taux d'alanine aminotransférase (ALAT) ou d'aspartate
aminotransférase (ASAT) sont supérieurs à 3 fois la
limite supérieure de la normale (LSN).
Le traitement
doit être arrêté temporairement si :
• ALAT
ou ASAT > 5 × LSN,
• un
patient développe une leucopénie (numération des globules blancs < 2 ×
109/L) ou une neutropénie (neutrophiles < 1 × 109/L), ou une lymphopénie
(lymphocytes < 0,2 × 109/L).
• un
patient présente une infection active et grave (c'est-à-dire nécessitant une
hospitalisation ou une prolongation d'hospitalisation).
Le traitement
peut être repris :
• après
normalisation des valeurs et sur la base d'une évaluation individuelle du
rapport bénéfice/risque.
En cas de
reprise du traitement, les transaminases hépatiques et la bilirubine totale
doivent être étroitement surveillées.
L'arrêt
définitif du traitement doit être envisagé si :
• ALAT
ou ASAT > 8 × LSN,
• ALAT
ou ASAT > 5 × LSN pendant plus de 2 semaines,
• ALAT
ou ASAT > 3 × LSN et bilirubine totale > 2 × LSN ou rapport international
normalisé (INR) > 1,5,
• ALAT
ou ASAT > 3 × LSN avec apparition de fatigue, nausées, vomissements, douleur
ou sensibilité dans le quadrant supérieur droit, fièvre, rash et/ou
éosinophilie (> 5 %),
• une
association entre l'avacopan et le dysfonctionnement
hépatique a été établie.
Populations particulières
Personnes âgées
Aucune adaptation posologique n'est nécessaire chez les patients âgés (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère ou modérée (voir rubrique Propriétés pharmacocinétiques).
L'avacopan n'a pas été étudié chez les sujets présentant une insuffisance hépatique sévère (classe C de Child-Pugh) et son utilisation n'est donc pas recommandée chez ces patients.
Insuffisance rénale
Aucune adaptation posologique n'est nécessaire en fonction de la fonction rénale (voir rubrique Propriétés pharmacocinétiques).
L'avacopan n'a pas été étudié chez les patients atteints de vascularite associée aux anticorps anti- cytoplasme des polynucléaires neutrophiles (ANCA) avec un débit de filtration glomérulaire estimé (DFGe) inférieur à 15 mL/min/1,73 m², sous dialyse, nécessitant une dialyse ou des échanges plasmatiques.
Maladie grave se manifestant par une hémorragie alvéolaire.
L'avacopan n'a pas été étudié chez les patients atteints d'une maladie grave se manifestant par une hémorragie alvéolaire.
Population pédiatrique
La sécurité et l'efficacité de l'avacopan chez les adolescents (âgés de 12 à 17 ans) n'ont pas encore été établies. Les données actuellement disponibles sont décrites aux rubriques Effets indésirables et Propriétés pharmacodynamiques mais aucune recommandation sur la posologie ne peut être donnée. La sécurité et l'efficacité de l'avacopan chez les enfants âgés de moins de 12 ans n'ont pas encore été établies. Aucune donnée n'est disponible.
Mode d'administration
Ce médicament doit être administré par voie orale.
Les gélules doivent être prises avec de la nourriture et avalées entières avec de l'eau. Elles ne doivent pas être écrasées, ni mâchées ni ouvertes.
Le pamplemousse et le jus de pamplemousse doivent être évités chez les patients traités par l'avacopan (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Durée de conservation :
4 ans
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation concernant la température. À conserver dans le flacon d'origine, à l'abri de la lumière.
Sans objet.
L'avacopan a été étudié chez des sujets sains à une dose quotidienne totale maximale de 200 mg (100 mg deux fois par jour) pendant 7 jours sans élément indiquant une toxicité limitant la dose. En cas de surdosage, il est recommandé de surveiller le patient afin de détecter tout signe ou symptôme d'effet indésirable et de recourir à un traitement symptomatique et des soins de support appropriés.
Classe pharmacothérapeutique : Inhibiteurs de complément, Code ATC : L04AJ05
Mécanisme d'action
L'avacopan est un antagoniste sélectif du récepteur 5a du
complément humain (C5aR1 ou CD88) ; il inhibe de manière compétitive
l'interaction entre le C5aR1 et l'anaphylatoxine C5a.
Le blocage spécifique et sélectif du C5aR1 par l'avacopan
réduit les effets pro-inflammatoires du C5a, qui comprennent l'activation, la
migration et l'adhérence des neutrophiles aux sites d'inflammation des petits
vaisseaux sanguins, la rétraction des cellules endothéliales et la perméabilité
vasculaires.
Effets pharmacodynamiques
L'avacopan bloque la régulation positive du CD11b (intégrine alpha M) induite par le C5a sur les neutrophiles prélevés chez des humains ayant reçu l'avacopan. La protéine CD11b facilite l'adhérence des neutrophiles aux surfaces endothéliales vasculaires, l'une des étapes du processus pathologique de la vascularite.
Efficacité et sécurité cliniques
Au total, 330 patients âgés de 13 ans ou plus atteints de granulomatose avec polyangéite (GPA) (54,8 %) ou de polyangéite microscopique (PAM) (45,2 %) ont été traités dans le cadre de l'étude ADVOCATE, une étude pivot de phase III, multicentrique, contrôlée contre comparateur actif, randomisée, en double aveugle et double placebo, menée sur 52 semaines.
Le plan de l'étude ADVOCATE est décrit dans la Figure 1.
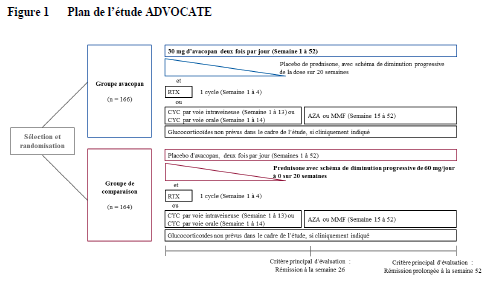
AZA = azathioprine ; CYC = cyclophosphamide ; MMF = mycophénolatemofétil ; RTX = rituximab
Les patients
ont été randomisés selon un rapport 1:1 dans l'un des deux groupes suivants :
• Groupe
avacopan (N = 166) : les patients ont reçu 30 mg d'avacopan deux fois par jour pendant 52 semaines, plus un
placebo équivalent à la prednisone, selon un schéma de diminution progressive
de la dose sur 20 semaines,
• Groupe
de comparaison (N = 164) : les patients ont reçu un placebo équivalent à l'avacopan deux fois par jour pendant 52 semaines plus de la
prednisone (schéma de diminution progressive de 60 mg/jour à 0 sur 20
semaines).
Tous les
patients des deux groupes ont reçu des traitements immunosuppresseurs standard
comprenant :
• du
rituximab à la dose de 375 mg/m² pour 4 doses
hebdomadaires par voie intraveineuse, ou
• du
cyclophosphamide par voie intraveineuse pendant 13
semaines (15 mg/kg jusqu'à 1,2 g toutes les 2 à 3 semaines), puis, à partir de
la semaine 15, azathioprine par voie orale à raison
de 1 mg/kg par jour avec augmentation progressive jusqu'à 2 mg/kg par jour (le mycophénolatemofétil à raison de
2 g par jour était autorisé à la place de l'azathioprine.
Si le mycophénolatemofétil
n'était pas toléré ou n'était pas disponible, le mycophénolate
sodique à enrobage entérique pouvait être administré à une dose cible de 1 440
mg/jour), ou
• du
cyclophosphamide par voie orale pendant 14 semaines
(2 mg/kg par jour), suivi d'azathioprine ou de mycophénolatemofétil/sodique par
voie orale à partir de la semaine 15 (même schéma posologique que le cyclophosphamide par voie intraveineuse).
Pour la première perfusion de rituximab, 100 mg de méthylprednisolone, ou équivalent, ont été administrés avant de commencer la perfusion de rituximab. Un pré-traitement par glucocorticoïdes était autorisé pour les deuxième, troisième et quatrième perfusions de rituximab.
Des réductions ou des ajustements de doses de cyclophosphamide, d'azathioprine et de mycophénolate ont été autorisés afin de se conformer aux approches standard visant à maximiser la sécurité de ces médicaments.
Le calendrier de diminution progressive des glucocorticoïdes suivant, prévu dans le protocole, a été utilisé (tableau 2).
Tableau 2 : Calendrier de diminution progressive des glucocorticoïdes - Dose de prednisone (mg par jour)
| Jour de l'étude | Avacopan | Comparateur | |
| Tous | ≥ 55 kg | < 55 kg | |
| 1 à 7 | 0 | 60 | 45 |
| 8 à 14 | 0 | 45 | 45 |
| 15 à 21 | 0 | 30 | 30 |
| 22 à 42 | 0 | 25 | 25 |
| 43 à 56 | 0 | 20 | 20 |
| 57 à 70 | 0 | 15 | 15 |
| 71 à 98 | 0 | 10 | 10 |
| 99 à 140 | 0 | 5 | 5 |
| ≥ 141 | 0 | 0 | 0 |
Les glucocorticoïdes non prévus dans le cadre de l'étude, à moins qu'ils ne soient strictement nécessaires en raison d'une affection nécessitant l'utilisation de glucocorticoïdes (telle qu'une insuffisance surrénalienne), devaient être évités autant que possible pendant l'étude. Cependant, les patients qui ont présenté une aggravation ou une rechute de leur vascularite associée aux ANCA pendant l'étude ont pu être traités avec un traitement limité de glucocorticoïdes.
Les patients
ont été stratifiés au moment de la randomisation pour obtenir un équilibre
entre les groupes de traitement sur la base de 3 facteurs :
• vascularite
associée aux ANCA nouvellement diagnostiquée ou récidivante,
• vascularite
associée aux ANCA positive à la protéinase 3 (PR3) ou à la myéloperoxydase
(MPO),
• traités
soit par du rituximab par voie intraveineuse, soit
par du cyclophosphamide par voie intraveineuse ou par
voie orale.
Les caractéristiques démographiques et pathologiques initiales des patients étaient similaires entre les deux groupes (tableau 3).
Tableau 3 : Caractéristiques initiales des patients sélectionnés dans l'étude pivot de phase III ADVOCATE (population en intention de traiter)
| Caractéristique démographique |
Avacopan
(N = 166) |
Comparateur
(N = 164) |
| Âge lors de la sélection | ||
| Moyenne (ET), années | 61 (14,6) | 61 (14,5) |
| Intervalle, années | 13-83 | 15-88 |
| Statut de la vascularite associée aux ANCA, n (%) | ||
| Nouvellement diagnostiquée | 115 (69,3) | 114 (69,5) |
| Rechute | 51 (30,7) | 50 (30,5) |
| Positivité aux ANCA, n (%) | ||
| PR3 | 72 (43,4) | 70 (42,7) |
| MPO | 94 (56,6) | 94 (57,3) |
| Type de vascularite associée aux ANCA, n (%) | ||
| Granulomatose avec polyangéite (GPA) | 91 (54,8) | 90 (54,9) |
| Polyangéite microscopique (PAM) | 75 (45,2) | 74 (45,1) |
| Score BVAS | ||
| Moyenne (ET) | 16,3 (5,87) | 16,2 (5,69) |
| DFGe | ||
| Moyenne (ET), mL/min/1,73 m² | 50,7 (30,96) | 52,9 (32,67) |
| Caractéristique démographique |
Avacopan (N = 166) |
Comparateur (N = 164) |
| Utilisation antérieure de glucocorticoïdes (lors de la sélection) | ||
| n (%) | 125 (75,3) | 135 (82,3) |
| Moyenne (ET), dose équivalente de prednisone (mg) | 907 (1 145,9) | 978 (1 157,5) |
ANCA =
anticorps anti-cytoplasme des polynucléaires neutrophiles ; BVAS = score
d'activité de la vascularite de Birmingham (Birmingham Vasculitis
Activity Score) ; MPO = myéloperoxydase ; PR3 =
protéinase 3,
ET =
écart-type.
Le but de l'étude était de déterminer l'efficacité de l'avacopan chez les patients atteints de vascularite associée aux ANCA, tout en permettant de réduire l'utilisation des glucocorticoïdes sans compromettre la sécurité ou l'efficacité.
L'objectif
principal était d'évaluer l'efficacité des traitements décrits ci-dessus pour
induire et maintenir une rémission chez les patients atteints de vascularite
associée aux ANCA en se basant sur les deux critères principaux suivants :
• proportion
de patients en rémission de la maladie, définie par l'obtention d'un score BVAS
de 0 et l'absence de prise de glucocorticoïdes pour le traitement de la
vascularite associée aux ANCA dans les 4 semaines précédant la Semaine 26,
• proportion
de patients en rémission prolongée, définie comme une rémission à la Semaine 26
sans rechute à la Semaine 52 et avec un score BVAS de 0 et ne prenant pas de
glucocorticoïdes pour le traitement de la vascularite associée aux ANCA dans
les 4 semaines précédant la Semaine 52.
Les deux critères principaux ont été testés de manière séquentielle pour la non-infériorité et la supériorité en utilisant une procédure de « gatekeeping » pour préserver l'erreur de type I à 0,05.
Les résultats de cette étude sont présentés dans le tableau 4.
Tableau 4 : Rémission à la Semaine 26 et rémission prolongée à la Semaine 52 dans l'étude pivot de phase III ADVOCATE (population en intention de traiter)
|
Avacopan
N = 166 n (%) |
Comparateur N = 164 n (%) |
Estimation de la différence de traitement en %a | |
| Rémission à la semaine 26 | 120 (72,3) | 115 (70,1) | 3,4 |
| IC à 95 % | 64,8 ; 78,9 | 62,5 ; 77,0 | -6,0 ; 12,8 |
| Rémission prolongée à la semaine 52 | 109 (65,7) | 90 (54,9) | 12,5 b |
| IC à 95 % | 57,9 ; 72,8 | 46,9 ; 62,6 | 2,6 ; 22,3 |
IC =
intervalle de confiance
a Les IC à 95 % bilatéraux ont été ajustés sur les facteurs de stratification de la randomisation.
b Valeurs de p de supériorité : 0,013 (bilatérales).
a Les IC à 95 % bilatéraux ont été ajustés sur les facteurs de stratification de la randomisation.
b Valeurs de p de supériorité : 0,013 (bilatérales).
L'efficacité observée était cohérente dans tous les sous-groupes pertinents, c'est-à-dire ceux présentant une maladie nouvellement diagnostiquée et une maladie en rechute, les ANCA PR3 et MPO positifs, la GPA et la PAM, et les hommes et les femmes. Les résultats d'efficacité par traitement sont présentés dans le tableau 5.
Tableau 5 : Rémission à la Semaine 26 et rémission prolongée à la Semaine 52 par traitement dans l'étude pivot de phase III ADVOCATE (population en intention de traiter)
|
Avacopan
n/N (%) |
Comparateur
n/N (%) |
Différence en %, IC à 95 %a |
|
| Rémission à la semaine 26 | |||
| Patients recevant du rituximab par voie intraveineuse | 83/107 (77,6) | 81/107 (75,7) | 1,9 (-9,5 ; 13,2) |
| Patients recevant du cyclophosphamide par voie intraveineuse ou voie orale | 37/59 (62,7) | 34/57 (59,6) | 3,1 (-14,7 ; 20,8) |
| Rémission prolongée à la semaine 52 | |||
| Patients recevant du rituximab par voie intraveineuse | 76/107 (71,0) | 60/107 (56,1) | 15,0 (2,2 ; 27,7) |
| Patients recevant du cyclophosphamide par voie intraveineuse ou orale | 33/59 (55,9) | 30/57 (52,6) | 3,3 (-14,8 ; 21,4) |
a Des intervalles de
confiance (IC) à 95 % bilatéraux sont calculés pour la différence de
proportions (avacopan moins comparateur) selon la
méthode de Wald.
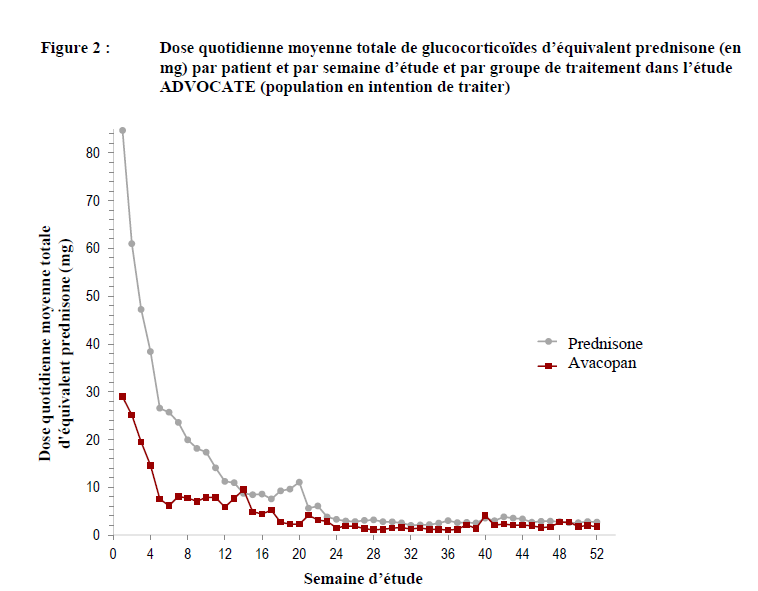
Toxicité des glucocorticoïdes
Dans l'étude pivot de phase III ADVOCATE, la dose cumulée totale moyenne d'équivalent prednisone, du Jour 1 à la fin du traitement, était environ 2,3 fois plus élevée dans le groupe de comparaison par rapport au groupe avacopan (3 846,9 mg vs 1 675,5 mg, respectivement).
De l'inclusion à la semaine 26, 86,1 % des patients utilisant l'avacopan ont reçu des glucocorticoïdes non prévus dans le cadre de l'étude. Dans le groupe de comparaison, la majorité des glucocorticoïdes utilisés était liée au traitement par la prednisone défini par le protocole.
L'indice de toxicité des glucocorticoïdes (GTI) évalue la morbidité liée aux glucocorticoïdes, y compris les mesures de l'indice de masse corporelle, la tolérance au glucose, les lipides, la myopathie stéroïdienne, la toxicité cutanée, la toxicité neuropsychiatrique et l'infection. Un GTI plus élevé indique une plus grande toxicité des glucocorticoïdes. Le GTI comprend le score d'aggravation cumulé (SAC) qui rend compte de la toxicité cumulée au cours du temps, et le score d'amélioration agrégé (SAA) qui rend compte à la fois de l'amélioration et de l'aggravation de la toxicité au cours du temps.
Les deux scores GTI (SAC et SAA) du groupe avacopan par rapport au groupe de comparaison sont résumés dans le tableau 6. Les mesures du GTI étaient des critères secondaires de l'étude et n'ont pas fait l'objet d'un ajustement dû à la multiplicité des tests.
Tableau 6 : Résultats de l'indice de toxicité des glucocorticoïdes dans l'étude pivot de phase III ADVOCATE (population en intention de traiter)
|
Avacopan
N = 166 |
Comparateur
N = 164 |
Différence entre les groupes,
IC à 95 % |
|
| Score d'aggravation cumulé (SAC) | |||
| Semaine 13 (moyenne des moindres carrés) | 25,7 | 36,6 | -11,0 (-19,7 ; -2,2) |
| Semaine 26 (moyenne des moindres carrés) | 39,7 | 56,6 | -16,8 (-25,6 ; -8,0) |
|
Avacopan
N = 166 |
Comparateur
N = 164 |
Différence entre les groupes,
IC à 95 % |
|
| Score d'amélioration agrégé (SAA) | |||
| Semaine 13 (moyenne des moindres carrés) | 9,9 | 23,2 | -13,3 (-22,2 ; -4,4) |
| Semaine 26 (moyenne des moindres carrés) | 11,2 | 23,4 | -12,1 (-21,1 ; -3,2) |
Population pédiatrique
Au total, 3
adolescents ont été inclus dans l'étude pivot de phase III ADVOCATE, deux dans
le groupe avacopan et un dans le groupe de
comparaison. Un adolescent du groupe avacopan a
interrompu le traitement en raison d'une aggravation de la vascularite rénale.
Le second patient adolescent ayant reçu l'avacopan a
terminé le traitement, a atteint la rémission à la Semaine 26 et la rémission
prolongée à la Semaine 52.
L'adolescente
du groupe de comparaison a arrêté le traitement en raison de la non-observance
de la contraception.
L'Agence Européenne des Médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec l'avacopan dans un ou plusieurs sous-groupes de la population pédiatrique dans le traitement de la vascularite associée aux ANCA (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Absorption
Lorsqu'il est administré sans nourriture, la concentration plasmatique maximale (Cmax) de l'avacopan survient après une durée médiane (tmax) d'environ 2 heures. Dans l'intervalle de doses de 10 à 30 mg, l'avacopan a montré une augmentation approximativement proportionnelle à la dose de l'exposition systémique.
L'administration de 30 mg en gélule au cours d'un repas riche en graisses et en calories augmente l'exposition plasmatique (ASC) de l'avacopan d'environ 72 % et retarde le tmax d'environ 3 heures ; cependant, la Cmax n'est pas modifiée.
Distribution
La liaison réversible aux protéines plasmatiques (par exemple, à l'albumine et à l'α1-glycoprotéine acide) de l'avacopan et du métabolite M1 est supérieure à 99,9 %. Le volume apparent de distribution est élevé (Vz/F 3 000 - 11 000 L), indiquant une large distribution tissulaire de la substance active.
Biotransformation
L'avacopan est éliminé principalement par le métabolisme de phase I. Après l'administration par voie orale d'avacopan radiomarqué, la majeure partie des matières liées à la substance active a été retrouvée dans les fèces sous forme de métabolites de phase I. Un métabolite circulant majeur (M1), un produit mono-hydroxylé de l'avacopan, était présent à hauteur d'environ 12 % de la totalité des matières liées à la substance active dans le plasma. Ce métabolite constitue 30 à 50 % de l'exposition de la molécule mère et a approximativement la même activité que l'avacopan sur le C5aR1. Le cytochrome P450 (CYP) 3A4 est la principale enzyme responsable de la clairance de l'avacopan et de la formation et de la clairance du métabolite M1.
L'interaction clinique entre l'avacopan et la simvastatine, substrat sensible du CYP3A4, est décrite dans la rubrique Interactions avec d'autres médicaments et autres formes d'interactions. L'avacopan est un inhibiteur faible du CYP2C9, comme l'indique l'augmentation modeste de l'ASC de la substance active, le célécoxib (1,15 fois).
In vitro, l'avacopan n'est pas un inhibiteur d'autres enzymes CYP et n'est pas un inducteur des enzymes CYP.
L'avacopan a montré une inhibition négligeable à faible des transporteurs courants in vitro. Par conséquent, les interactions cliniquement pertinentes sont peu probables quand l'avacopan est administré de façon concomitante avec des substances qui sont des substrats ou des inhibiteurs de ces transporteurs.
Élimination
Selon l'analyse pharmacocinétique de population, la clairance corporelle apparente totale (CL/F) de l'avacopan est de 16,3 L/h (IC à 95 % : 13,1 - 21,1 L/h). La demi-vie d'élimination terminale médiane est de 510 heures (21 jours) d'après l'analyse pharmacocinétique de population. Lorsque l'avacopan est arrêté après que l'état d'équilibre a été atteint, la concentration plasmatique résiduelle de l'avacopan devrait diminuer pour atteindre ~20 %, < 10 % et < 5 % de la concentration maximale de l'état d'équilibre environ 4 semaines, 7 semaines et 10 semaines, respectivement, après l'administration de la dernière dose.
Après l'administration par voie orale d'avacopan radiomarqué, environ 77 % et 10 % de la radioactivité a été retrouvée dans les selles et les urines, respectivement, et 7 % et moins de 0,1 % de la dose radioactive a été retrouvée sous forme d'avacopan inchangé dans les selles et les urines, respectivement. Ces résultats suggèrent que la principale voie de clairance de l'avacopan est le métabolisme suivi de l'excrétion biliaire des métabolites dans les selles, et que l'excrétion directe de l'avacopan dans les urines ou les selles par la bile est négligeable.
Populations particulières
Personnes âgées
L'analyse pharmacocinétique de population n'a pas mis en évidence d'effet significatif de l'âge (chez les adultes) sur l'exposition plasmatique de l'avacopan ; cependant, les données pharmacocinétiques chez les patients âgés de plus de 75 ans dans les études cliniques sont limitées. Aucune adaptation posologique n'est nécessaire chez les patients âgés (voir rubrique Posologie et mode d'administration).
Insuffisance hépatique
Les propriétés pharmacocinétiques de l'avacopan ont été évaluées chez 16 patients présentant une insuffisance hépatique légère (classe A de Child-Pugh) ou modérée (classe B de Child-Pugh). En comparaison avec les témoins normaux, aucune différence pharmacologiquement pertinente dans l'exposition (rapports moyens de la Cmax et de l'ASC ≤ 1,3) de l'avacopan ou de son principal métabolite M1 n'a été observée ; par conséquent, aucune adaptation posologique n'est nécessaire (voir rubrique Posologie et mode d'administration).
L'avacopan n'a pas été étudié chez les patients présentant une insuffisance hépatique sévère (classe C de Child-Pugh) (voir rubrique Posologie et mode d'administration).
Insuffisance rénale
D'après l'analyse pharmacocinétique de population, l'exposition plasmatique de l'avacopan est similaire entre les patients présentant une insuffisance rénale et les sujets sains. Par conséquent, aucune adaptation posologique n'est nécessaire en fonction de la fonction rénale (voir rubrique Posologie et mode d'administration).
L'avacopan n'a pas été étudié chez les patients atteints de vascularite associée aux ANCA avec un DFGe inférieur à 15 mL/min/1,73 m², sous dialyse, nécessitant une dialyse ou des échanges plasmatiques.
Tavneos n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration répétée, génotoxicité ou cancérogenèse n'ont pas révélé de risque particulier pour l'Homme.
Fertilité et développement embryonnaire précoce
L'avacopan n'a produit aucun effet sur la fonction reproductive (fertilité) ou le développement précoce des mâles et des femelles chez les hamsters à des doses orales équivalentes à 6,8 fois l'ASC clinique.
Développement embryonnaire et fœtal
L'avacopan ne s'est pas révélé tératogène lorsqu'il a été administré par voie orale à des hamsters et des lapins. Chez les hamsters, une incidence accrue de variations squelettiques (côte surnuméraire thoracolombaire courte) a été observée à une exposition équivalente à 5,3 fois l'ASC clinique. Chez le lapin, l'avacopan a provoqué une toxicité maternelle (signes cliniques indésirables et avortements), mais aucune toxicité fœtale à une exposition équivalente à 0,6 fois l'ASC clinique.
Développement prénatal et postnatal
L'avacopan
n'a pas entraîné d'effets indésirables chez la progéniture femelle
lorsqu'il a été administré à des hamsters à des expositions allant
jusqu'à 6,3 fois l'ASC clinique pendant la gestation et tout au long de
la lactation jusqu'au sevrage. Chez les mâles, on a observé un léger
retard dans la séparation préputiale à une exposition de 3,7 fois l'ASC
clinique. Cette observation isolée a été considérée comme ayant une
faible signification toxicologique et n'a pas été associée à une
altération de la performance reproductive.
L'analyse des taux plasmatiques d'avacopan chez les mères allaitantes
et des taux plasmatiques chez la progéniture allaitée a révélé la
présence d'avacopan, suggérant que l'avacopan est probablement sécrété
dans le lait des hamsters en lactation.
Carcinogénicité
Le potentiel cancérigène de l'avacopan a été évalué dans une étude de 2 ans chez les rats et les hamsters.
Chez les rats mâles traités par l'avacopan, une incidence légèrement
accrue d'adénome thyroïdien à cellules C a été notée ; cette
augmentation n'était pas statistiquement significative et l'incidence
se situait dans l'intervalle des témoins historiques. L'avacopan ne
s'est pas révélé cancérigène chez les hamsters, espèce
pharmacologiquement pertinente.
Pas d'exigences particulières.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste
I.
Médicament
nécessitant une surveillance particulière pendant le traitement.
Prescription
initiale hospitalière annuelle.
Prescription
réservée aux spécialistes et services DERMATOLOGIE.
Prescription
réservée aux spécialistes et services MEDECINE INTERNE.
Prescription
réservée aux spécialistes et services NEPHROLOGIE.
Prescription
réservée aux spécialistes et services PNEUMOLOGIE.
Prescription
réservée aux spécialistes et services RHUMATOLOGIE.
Renouvellement non restreint.
Gélule
Gélules avec un corps jaune et une coiffe orange clair portant la mention « CCX168 » imprimée à l'encre noire.
Une gélule a une longueur de 22 mm et un diamètre de 8 mm (taille 0).
Flacon en polyéthylène haute densité (PEHD) avec fermeture de sécurité enfant et scellage par induction.
Boîte de 30 gélules.
Chaque gélule contient 10 mg d'avacopan.
Excipient à effet notoire :
Chaque gélule contient 245 mg d'hydroxystéarate de macrogolglycérol.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Contenu de la gélule
Hydroxystéarate de macrogolglycérol
Macrogol (4000)
Enveloppe de la gélule
Gélatine
Oxyde de fer rouge (E172)
Oxyde de fer jaune (E172)
Dioxyde de titane (E171)
Polysorbate 80
Encre imprimée
Oxyde de fer noir (E172)
Gomme-laque
Hydroxyde de potassium