
SPEVIGO 450 mg, solution à diluer pour perfusion, boîte de 2 flacons de 7,50 ml
Dernière révision : 25/09/2024
Taux de TVA : 2.1%
Laboratoire exploitant : BOEHRINGER INGELHEIM FRANCE
Source :

Spevigo est indiqué dans le traitement des poussées de psoriasis pustuleux généralisé (PPG) chez les adultes et les adolescents à partir de 12 ans, en monothérapie.
Hypersensibilité sévère ou susceptible de menacer le pronostic vital à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients (voir rubrique Mises en garde spéciales et précautions d'emploi).
Infections actives cliniquement significatives (p. ex., tuberculose active ; voir rubrique Mises en garde spéciales et précautions d'emploi).
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Infections
Le spésolimab peut accroître le risque d'infections (voir rubrique Effets indésirables).
Chez les patients présentant une infection chronique ou des antécédents d'infection récidivante, il convient de tenir compte des risques potentiels et des bénéfices cliniques attendus du traitement avant de prescrire le spésolimab. En cas d'infection active cliniquement significative, le traitement par spésolimab ne doit pas être instauré avant que l'infection ait disparu ou soit traitée de façon adéquate. Les patients doivent être informés de la nécessité de consulter un médecin si des signes ou symptômes évocateurs d'une infection cliniquement significative surviennent après le traitement par spésolimab.
Dépistage de la tuberculose avant le traitement
Le dépistage de la tuberculose doit être effectué avant l'instauration du traitement par spésolimab. Le spésolimab est contre-indiqué chez les patients présentant une tuberculose active (voir rubrique Contre-indications).
Un traitement antituberculeux doit être envisagé avant l'instauration du traitement par spésolimab chez les patients présentant une tuberculose latente ou des antécédents de tuberculose, ou ayant pu être exposés à des personnes présentant une tuberculose active, si l'administration d'un traitement complet et approprié ne peut être confirmée. Après le traitement par spésolimab, les patients doivent être placés sous surveillance à la recherche de signes et symptômes de tuberculose active.
Hypersensibilité et réactions liées à la perfusion
Une hypersensibilité ou des réactions liées à la perfusion peuvent survenir avec les anticorps monoclonaux tels que le spésolimab. L'hypersensibilité peut se manifester par des réactions immédiates telles qu'une anaphylaxie, ou par des réactions plus tardives telles qu'un syndrome d'hypersensibilité médicamenteuse avec éosinophilie et symptômes systémiques (syndrome DRESS).
Si le patient présente des signes d'anaphylaxie ou d'autre hypersensibilité grave, le traitement par spésolimab doit être immédiatement arrêté et un traitement approprié doit être instauré (voir rubrique Contre-indications).
En cas de réaction d'hypersensibilité d'intensité légère ou modérée durant une perfusion intraveineuse ou d'autres réactions liées à la perfusion, le traitement doit être arrêté et l'instauration d'un traitement médical approprié doit être envisagée (p. ex., antihistaminiques systémiques et/ou corticostéroïdes). À la résolution de la réaction, la perfusion pourra être reprise à un débit plus lent qui sera augmenté progressivement jusqu'à la fin de la perfusion (voir rubrique Posologie et mode d'administration).
Utilisation chez les patients présentant une poussée de PPG engageant le pronostic vital
Il n'existe aucune donnée sur l'utilisation du spésolimab chez les patients présentant une poussée de PPG engageant le pronostic vital ou une poussée nécessitant une prise en charge en soins intensifs.
Utilisation concomitante avec d'autres traitements du PPG
La sécurité et l'efficacité du spésolimab en association avec des immunosuppresseurs, y compris des traitements biologiques, n'ont pas été évaluées de manière systématique (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). Dans l'étude clinique menée sur le traitement des poussées de PPG, une période de sevrage thérapeutique (wash-out) a été instaurée pour la plupart des autres traitements (produits biologiques, autres traitements immunomodulateurs systémiques), tandis que d'autres traitements ont été arrêtés avant l'instauration du traitement par spésolimab sans qu'une période de sevrage thérapeutique (wash-out) soit requise (méthotrexate, cyclosporine, rétinoïdes, traitements topiques) (voir rubrique Propriétés pharmacodynamiques). L'utilisation concomitante d'autres immunosuppresseurs et de spésolimab n'est pas recommandée. Au moment de l'instauration du traitement par spésolimab, les autres traitements du PPG doivent être arrêtés et d'autres traitements (p. ex. par immunosuppresseurs systémiques) ne doivent pas être utilisés de manière concomitante pour traiter la poussée.
Retraitement
Des données d'efficacité et de sécurité
très limitées sont disponibles sur le retraitement par spésolimab pour une
nouvelle poussée ultérieure. Dans l'essai Effisayil 1, cinq patients ont reçu
une nouvelle dose pour le traitement d'une nouvelle poussée et ont été suivis
pendant 8 semaines minimum.
Vaccins
On ignore si le spésolimab affecte l'efficacité des vaccins.
Aucune donnée n'est disponible sur la transmission secondaire potentielle d'une infection par des vaccins vivants chez les patients recevant du spésolimab (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). L'intervalle entre l'administration d'un vaccin vivant et l'instauration du traitement par spésolimab doit être d'au moins 4 semaines. Les vaccins vivants ne doivent pas être administrés pendant au moins 16 semaines après le traitement par spésolimab.
Pour plus d'informations concernant l'administration de vaccins avant l'instauration du traitement pour la prévention des poussées de PPG, se référer au résumé des caractéristiques du produit de Spevigo 150 mg solution injectable en seringue préremplie.
Neuropathie périphérique
On ignore si le spésolimab est associé à un risque de neuropathie périphérique. Des cas de neuropathie périphérique ont été signalés lors des essais cliniques menés avec le spésolimab. Les médecins doivent être attentifs à l'apparition de symptômes pouvant évoquer la survenue d'une neuropathie périphérique.
Excipients
Ce médicament contient moins de 1 mmol (23 mg) de sodium par dose, c.-à-d. qu'il est essentiellement « sans sodium ».
Résumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés sont les infections (17,1 %), dont l'infection des voies urinaires considérée comme grave chez 1 patient (2,9 %) (voir « Description de certains effets indésirables »).
Liste tabulée des effets indésirables
Le tableau 1 présente la liste des effets indésirables rapportés lors des essais cliniques. Les effets indésirables sont répertoriés par classe de systèmes d'organes MedDRA et par catégorie de fréquence selon la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Tableau 1 Effets indésirables
|
Classe de système d'organe |
Effet indésirable |
Fréquence |
|
Infections et infestations |
Infectiona |
Très fréquent |
|
Affections de la peau et du tissu souscutané |
Prurit |
Fréquent |
|
Troubles généraux et anomalies au site d'administration |
Réactions au site d'injection |
Très fréquentb |
|
Fatigue |
Fréquent |
a Les infections les plus fréquemment rapportées étaient les infections des
voies urinaires (Fréquent) et les infections des voies aériennes supérieures
(Très fréquent).
b Non signalé dans l'essai Effisayil 1
Description de certains effets indésirables
Infections
Durant la période contrôlée contre placebo d'une semaine de l'essai Effisayil 1, des infections ont été rapportées chez 17,1 % des patients traités par spésolimab, contre 5,6 % des patients ayant reçu le placebo. Dans l'essai Effisayil 1, une infection grave (infection des voies urinaires) a été rapportée chez 1 patient (2,9 %) du groupe spésolimab et chez aucun patient du groupe placebo. Durant la période contrôlée contre placebo de 48 semaines maximum de l'essai Effisayil 2, des infections ont été rapportées chez 33,3 % des patients traités par Spevigo et chez 33,3 % des patients ayant reçu le placebo. Dans l'essai Effisayil 2, des infections graves ont été rapportées chez 3 patients (3,2 %) du groupe Spevigo et chez aucun patient du groupe placebo.
Les infections observées au cours des essais cliniques sur le spésolimab étaient généralement d'intensité légère à modérée, sans profil spécifique concernant l'agent pathogène ou la nature de l'infection.
Réactions au site d'injection
Les réactions au site d'injection comprennent : érythème, gonflement, douleur, induration, chaleur, desquamation, papule, prurit, éruption cutanée et urticaire au site d'injection. Les réactions au site d'injection étaient généralement d'intensités légères à modérées.
Population pédiatrique
Les données disponibles chez les adolescents sont limitées. Huit (8) patients adolescents atteints de PPG âgés de 14 à 17 ans ont été inclus dans l'essai Effisayil 2 (voir rubrique Propriétés pharmacodynamiques). Dans l'ensemble, le profil de sécurité chez les adolescents traités avec le spésolimab (n = 6) correspondait à celui des adultes et aucun nouveau problème de sécurité n'a été identifié.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
AVANT
INSTAURATION du traitement :
- EFFECTUER un dépistage de la tuberculose.
SURVEILLANCE du traitement :
- Rechercher des signes et symptômes de tuberculose active.
ALLAITEMENT :
- L'excrétion d'anticorps IgG dans le lait s'observe durant les premiers jours après l'accouchement et diminue rapidement par la suite jusqu'à atteindre de faibles concentrations. En conséquence, des anticorps IgG peuvent être transmis au nouveau-né durant les premiers jours de l'allaitement. Durant cette courte période, un risque pour l'enfant allaité ne peut être exclu. Passé cette période, le spésolimab peut être utilisé pendant l'allaitement si cela est cliniquement nécessaire. Si le traitement a été arrêté avant le dernier trimestre de grossesse, l'allaitement peut être commencé immédiatement après l'accouchement.
- EFFECTUER un dépistage de la tuberculose.
SURVEILLANCE du traitement :
- Rechercher des signes et symptômes de tuberculose active.
ALLAITEMENT :
- L'excrétion d'anticorps IgG dans le lait s'observe durant les premiers jours après l'accouchement et diminue rapidement par la suite jusqu'à atteindre de faibles concentrations. En conséquence, des anticorps IgG peuvent être transmis au nouveau-né durant les premiers jours de l'allaitement. Durant cette courte période, un risque pour l'enfant allaité ne peut être exclu. Passé cette période, le spésolimab peut être utilisé pendant l'allaitement si cela est cliniquement nécessaire. Si le traitement a été arrêté avant le dernier trimestre de grossesse, l'allaitement peut être commencé immédiatement après l'accouchement.
Grossesse
Il n'existe pas de données ou il existe des données limitées sur l'utilisation du spésolimab chez la femme enceinte. Les études non cliniques menées avec un substitut d'anticorps monoclonal antiIL36R murin n'ont pas mis en évidence d'effets délétères directs ou indirects sur la reproduction (voir rubrique Données de sécurité préclinique). L'immunoglobuline humaine (IgG) est connue pour traverser la barrière placentaire. Par mesure de précaution, il est préférable d'éviter l'utilisation du spésolimab pendant la grossesse.
Allaitement
Il n'existe pas de données sur l'excrétion du spésolimab dans le lait maternel. Chez l'être humain, l'excrétion d'anticorps IgG dans le lait s'observe durant les premiers jours après l'accouchement et diminue rapidement par la suite jusqu'à atteindre de faibles concentrations. En conséquence, des anticorps IgG peuvent être transmis au nouveau-né durant les premiers jours de l'allaitement. Durant cette courte période, un risque pour l'enfant allaité ne peut être exclu. Passé cette période, le spésolimab peut être utilisé pendant l'allaitement si cela est cliniquement nécessaire. Si le traitement a été arrêté avant le dernier trimestre de grossesse, l'allaitement peut être commencé immédiatement après l'accouchement.
Fertilité
Aucune donnée n'est disponible concernant les effets du spésolimab sur la fertilité humaine. Les études menées sur la souris avec un substitut d'anticorps monoclonal anti-IL36R murin n'ont pas mis en évidence d'effets délétères directs ou indirects sur la fertilité par antagonisme de l'IL36R (voir rubrique Données de sécurité préclinique).
Aucune étude d'interaction n'a été réalisée. Chez les patients atteints de PPG, il n'est pas attendu que le spésolimab ait un impact sur les médicaments concomitants métabolisés par les enzymes du CYP.
Aucun vaccin vivant ne doit être co-administré avec le spésolimab (voir rubrique Mises en garde spéciales et précautions d'emploi).
L'expérience de l'utilisation concomitante du spésolimab avec des immunosuppresseurs chez les patients atteints de PPG est limitée (voir rubrique Mises en garde spéciales et précautions d'emploi).
Le traitement doit être instauré et supervisé par un médecin expérimenté dans la prise en charge des maladies inflammatoires cutanées.
Le traitement peut être initié en injection sous-cutanée avec la seringue préremplie pour la prévention des poussées de PPG (se référer au résumé des caractéristiques du produit de Spevigo 150 mg solution injectable en seringue préremplie), ou avec une dose de spésolimab par voie intraveineuse en traitement d'une poussée de PPG.
Posologie
La dose recommandée pour le traitement d'une poussée de PPG chez les adultes et les adolescents âgés de 12 ans ou plus et pesant au moins 40 kg est de 900 mg (deux flacons de 450 mg) en dose unique administrée en perfusion intraveineuse. Si les symptômes de la poussée persistent, une dose supplémentaire de 900 mg peut être administrée 1 semaine après la première dose.
Spevigo n'a pas été étudié chez les patients pesant moins de 40 kg. D'après les modélisations et les simulations pharmacocinétiques, la dose recommandée chez les adolescents âgés de 12 ans ou plus et pesant ≥ 30 et < 40 kg est une dose unique de 450 mg (un flacon de 450 mg) administrée en perfusion intraveineuse (voir rubrique Propriétés pharmacocinétiques). Si les symptômes de la poussée persistent, une dose supplémentaire de 450 mg (un flacon de 450 mg) peut être administrée 1 semaine après la première dose.
Les données cliniques concernant le traitement des poussées ultérieures sont très limitées (voir rubrique Mises en garde spéciales et précautions d'emploi).
Les données cliniques concernant
l'utilisation concomitante d'autres traitements du PPG avec le spésolimab sont limitées.
Le spésolimab ne doit pas être utilisé avec d'autres traitements du PPG, tels
que les immunosuppresseurs systémiques, pour traiter une poussée (voir
rubriques Mises en garde spéciales et précautions d'emploi et Interactions avec d'autres médicaments et autres formes d'interactions).
Populations particulières
Patients âgés
Aucun ajustement posologique n'est requis.
Insuffisance rénale ou hépatique
Le spésolimab n'a pas été étudié spécifiquement dans ces populations de patients. Ces troubles n'ont généralement pas d'incidence cliniquement significative sur la pharmacocinétique des anticorps monoclonaux, et aucun ajustement posologique n'est jugé nécessaire.
Population pédiatrique
La sécurité et l'efficacité du spésolimab chez les enfants âgés de moins de 12 ans n'ont pas été établies. Aucune donnée n'est disponible.
Mode d'administration
Ce médicament doit être administré exclusivement par perfusion intraveineuse. Il ne doit pas être administré en injection intraveineuse rapide ni en bolus.
Après dilution dans une solution injectable de chlorure de sodium à 9 mg/mL (0,9 %), le médicament doit être administré en perfusion intraveineuse continue au moyen d'une tubulure munie d'un filtre intégré stérile, apyrogène et à faible liaison protéique (pores de 0,2 micron) sur 90 minutes. Aucune autre perfusion ne doit être administrée en parallèle par le même accès veineux.
En cas de ralentissement ou d'interruption temporaire de la perfusion, la durée totale de la perfusion (en tenant compte de la durée de l'interruption) ne doit pas excéder 180 minutes (voir rubrique Mises en garde spéciales et précautions d'emploi).
Pour les instructions concernant la dilution du médicament avant administration, voir la rubrique Précautions particulières d’élimination et de manipulation.
Durée de conservation :
Flacon non-ouvert
3 ans.
Après ouverture
D'un point de vue microbiologique, le médicament doit être dilué et administré immédiatement après ouverture du flacon.
Après préparation de la perfusion
La solution diluée présente une stabilité physico-chimique en cours d'utilisation pendant une période de 24 heures à une température comprise entre 2 °C et 30 °C.
D'un point de vue microbiologique, la solution pour perfusion diluée doit être utilisée immédiatement. Dans le cas contraire, les conditions de conservation après dilution relèvent de la responsabilité de l'utilisateur, et la durée de conservation ne doit normalement pas dépasser 24 heures à une température comprise entre 2 °C et 8 °C, sauf si la dilution a été réalisée dans des conditions d'asepsie contrôlées et validées. Entre le moment de la préparation et le début de l'administration, la solution pour perfusion doit être protégée de la lumière, conformément aux procédures locales standard.
Précautions particulières de conservation :
À conserver au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler.
À conserver dans l'emballage d'origine à l'abri de la lumière.
Avant utilisation, le flacon non ouvert peut être conservé à des températures ne dépassant pas 30 °C pendant 24 heures maximum dès lors qu'il est conservé dans son emballage d'origine à l'abri de la lumière.
Pour les conditions de conservation du médicament après ouverture et dilution, voir la rubrique Durée de conservation.
Ce médicament ne doit pas être mélangé avec d'autres médicaments à l'exception de ceux mentionnés dans la rubrique Précautions particulières d'élimination et de manipulation.
La dose maximale de spésolimab administrée dans le cadre des essais cliniques était de 1 200 mg par voie intraveineuse ou sous-cutanée. Les effets indésirables observés après l'administration d'une dose unique ou de doses répétées pouvant aller jusqu'à 1 200 mg concordaient avec le profil de sécurité connu du spésolimab.
En cas de surdosage, il est recommandé de surveiller l'apparition de signes ou symptômes d'effets indésirables et d'instaurer un traitement symptomatique si nécessaire.
Classe pharmacothérapeutique : immunosuppresseurs, inhibiteurs d'interleukines, Code ATC :
L04AC22
Mécanisme d'action
Le spésolimab est un anticorps monoclonal antagoniste humanisé de type immunoglobuline G1 (IgG1) qui bloque la voie de signalisation du récepteur humain de l'interleukine 36 (IL36R). La liaison du spésolimab à l'IL36R empêche l'activation de l'IL36R par ses ligands (IL36 α, β et ?) ainsi que l'activation en aval des voies de signalisation pro-inflammatoires.
Effets pharmacodynamiques
Après l'administration intraveineuse de spésolimab à des patients atteints de PPG, une baisse des taux de protéine C réactive (CRP), d'IL6, de cytokines produites par les lymphocytes T auxiliaires (Th1/Th17), de marqueurs d'inflammation produits par les kératinocytes, de médiateurs neutrophiles et de cytokines pro-inflammatoires a été observée dans le sérum et au niveau de la peau à la semaine 1 par rapport à l'inclusion, et a été associée à une baisse de la sévérité clinique. Lors de la dernière mesure à la semaine 8 de l'essai Effisayil 1, la réduction de ces biomarqueurs s'était accentuée.
Efficacité et sécurité cliniques
Effisayil 1 (1368-0013)
Une étude randomisée, en double aveugle, contrôlée contre placebo (Effisayil 1) a été menée pour évaluer l'efficacité et la sécurité cliniques du spésolimab chez des patients adultes présentant des poussées de PPG diagnostiquées selon les critères ERASPEN (European Rare And Severe Psoriasis Expert Network, réseau européen de spécialistes des formes rares et sévères de psoriasis), quel que soit le statut mutationnel IL36RN. Les patients étaient randomisés s'ils avaient présenté une poussée de PPG d'intensité modérée à sévère, définie par un score total GPPGA (Generalised Pustular Psoriasis Physician Global Assessment, évaluation globale du psoriasis pustuleux généralisé par le médecin) d'au moins 3 (atteinte modérée) (sur une échelle de 0 [absence de lésion cutanée] à 4 [atteinte sévère]), par la présence de pustules récentes (pustules nouvellement apparues ou aggravation de pustules existantes), par un sous-score de pustulation d'au moins 2 (atteinte légère) à l'échelle GPPGA et par la présence d'un érythème et de pustules sur au moins 5 % de la surface corporelle. Les patients ont dû arrêter leurs traitements systémiques et topiques du PPG avant la randomisation (voir le tableau 2). Les patients qui présentaient une poussée de PPG engageant le pronostic vital ou nécessitant une prise en charge en soins intensifs ont été exclus de l'étude.
Tableau 2 : Intervalle minimum entre l'arrêt des médicaments dont l'usage est restreint pour le traitement du PPG et la randomisation (Effisayil 1)*
|
Durée de la période de sevrage thérapeutique (wash-out) |
Médicaments ou classe de médicaments |
|
2 mois |
adalimumab, alemtuzumab, briakinumab, brodalumab, éfalizumab, guselkumab, infliximab, ixékizumab, natalizumab, risankizumab, rituximab, sécukinumab, tildrakizumab, ustékinumab, visilizumab, produits expérimentaux contre le psoriasis (non biologiques) |
|
6 semaines |
étanercept |
|
30 jours |
traitements immunomodulateurs systémiques (p. ex. corticostéroïdes**, cyclophosphamide), tofacitinib, aprémilast ; autres traitements systémiques du psoriasis (p. ex. fumarates) ; tout dispositif ou médicament expérimental (à l'exclusion des produits destinés à traiter le psoriasis) ; photochimiothérapie (p. ex. puvathérapie) ; aphérèse par adsorption de granulocytes et de monocytes |
|
7 jours |
anakinra |
* Traitements dont l'instauration était interdite 1 semaine avant la randomisation : photothérapie (p. ex. UVA, UVB), traitement topique du psoriasis ou d'autres affections cutanées (p. ex. corticostéroïdes topiques, analogues de la vitamine D topique, goudron, anthraline, rétinoïdes topiques) ; traitements dont l'instauration était interdite 2 semaines avant la randomisation, dont la dose ne pouvait être augmentée dans les 2 semaines précédant la randomisation et qui devaient être arrêtés avant la première administration : méthotrexate, cyclosporine, rétinoïdes.
** Aucune restriction pour les corticostéroïdes inhalés utilisés dans le traitement de l'asthme ou les corticostéroïdes administrés sous forme de gouttes dans l'œil ou l'oreille.
Le critère d'évaluation principal de l'étude était la proportion de patients présentant un sous-score de pustulation de 0 (absence de pustules visibles) à l'échelle GPPGA à la semaine 1 post-traitement. Le principal critère d'évaluation secondaire était la proportion de patients présentant un score total de 0 ou 1 (absence ou quasi-absence de lésions cutanées) à l'échelle GPPGA à la semaine 1. Pour le sousscore de pustulation de 0 à l'échelle GPPGA et le score total de 0/1 à l'échelle GPPGA, la méthode d'imputation des non-répondeurs a été utilisée en cas de recours à un médicament de rattrapage (traitement laissé au choix de l'investigateur en cas d'aggravation de la maladie) ou de secours (dose unique de 900 mg de spésolimab par voie intraveineuse) et pour les données manquantes.
Au total, 53 patients ont été randomisés (selon un rapport de 2:1) pour recevoir une perfusion intraveineuse unique de 900 mg de spésolimab (n = 35) ou de placebo (n = 18). Les patients des deux groupes qui présentaient toujours des symptômes de poussée à la semaine 1 pouvaient recevoir une perfusion intraveineuse unique de 900 mg de spésolimab en ouvert : 12 patients (34 %) du bras spésolimab ont reçu une seconde dose de spésolimab, et 15 patients (83 %) du bras placebo ont reçu une dose de spésolimab au jour 8. Par ailleurs, 6 patients (4 dans le bras spésolimab et 2 dans le bras placebo) ont reçu une perfusion intraveineuse unique de 900 mg de spésolimab en traitement aigu d'une récidive de poussée après le jour 8.
La population de l'étude comptait 32 % d'hommes et 68 % de femmes. L'âge moyen était de 43 ans (intervalle : 21 à 69) ; 55 % des patients étaient caucasiens et 45 % étaient asiatiques. La majorité des patients inclus dans l'étude présentaient un sous-score de pustulation de 3 (43 %) ou 4 (36 %) à l'échelle GPPGA et un score total de 3 (81 %) ou 4 (19 %) à l'échelle GPPGA. Dans 24,5 % des cas, les patients avaient préalablement reçu un traitement biologique pour leur PPG.
Critère d'évaluation principal et principal critère d'évaluation secondaire d'efficacitéÀ la semaine 1, une différence statistiquement significative de la proportion de patients présentant un sous-score de pustulation de 0 (absence de pustules visibles) à l'échelle GPPGA et un score total de 0 ou 1 (absence ou quasi-absence de lésions cutanées) à l'échelle GPPGA a été observée entre le bras spésolimab et le bras placebo (voir tableau 3).
Tableau 3 : Sous-score de pustulation à l'échelle GPPGA et score total à l'échelle GPPGA à la semaine 1 (Effisayil 1)
|
|
Placebo |
|
spésolimab 900 mg en IV |
|
Nombre de patients analysés |
18 |
|
35 |
|
Patients présentant un sous-score de pustulation de 0 à l'échelle GPPGA, n (%) |
1 (5,6) |
|
19 (54,3) |
|
Valeur p* |
|
0,0004 |
|
|
Patients présentant un score total de 0 ou 1 à l'échelle GPPGA, n (%) |
2 (11,1) |
15 (42,9) |
|
|
Valeur p* |
|
0,0118 |
|
GPPGA : Generalised Pustular Psoriasis Physician Global Assessment (évaluation globale du psoriasis pustuleux généralisé par le médecin) ; IV : intraveineuse * Valeur p unilatérale
Que ce soit pour le critère d'évaluation principal ou le principal critère d'évaluation secondaire, un effet thérapeutique a été observé chez tous les patients, quel que soit leur statut mutationnel IL36RN.
Effisayil 2 (1368-0027)
Une étude de phase IIb randomisée, en double aveugle, contrôlée contre placebo (Effisayil 2) a évalué l'efficacité et la sécurité du spésolimab en administration sous-cutanée chez des patients adultes et adolescents présentant des antécédents de PPG diagnostiqué selon les critères ERASPEN, quel que soit leur statut mutationnel IL36RN, et ayant déjà présenté au moins deux poussées de PPG d'intensité modérée à sévère. Les patients étaient randomisés s'ils présentaient un score total de 0 ou 1 à l'échelle GPPGA au moment de la sélection et de la randomisation. Les patients devaient arrêter tout traitement systémique et topique du PPG avant ou au moment de la randomisation. Ces patients devaient avoir présentés des antécédents de poussées de PPG sous traitement ou lors d'une diminution de dose ou lors de l'interruption de ces traitements concomitants.
Le critère principal de l'étude était le délai de survenue de la première poussée de PPG jusqu'à la semaine 48 (définie comme un sous-score de pustulation ≥ 2 à l'échelle GPPGA et une augmentation ≥ 2 du score total GPPGA par rapport à l'inclusion). Le principal critère secondaire était la survenue d'au moins une poussée de PPG jusqu'à la semaine 48. Les autres critères secondaires à la semaine 48 étaient le délai de survenue de la première aggravation à l'échelle PSS (Psoriasis Symptom Scale, échelle d'évaluation des symptômes de psoriasis) et au questionnaire DLQI (Dermatology Quality of Life Index, questionnaire d'évaluation de la qualité de vie en dermatologie), définie comme une augmentation de 4 points du score total par rapport à l'inclusion.
Au total, 123 patients ont été randomisés (rapport de 1:1:1:1) pour recevoir l'un des 4 traitements de l'étude (voir tableau 4).
Tableau 4 : Groupes de traitement de l'essai Effisayil 2
|
|
Dose de charge |
Doses suivantes |
|
spésolimab |
600 mg par voie sous-cutanée |
300 mg par voie sous-cutanée toutes les 4 semaines |
|
spésolimab |
600 mg par voie sous-cutanée |
300 mg par voie sous-cutanée toutes les 12 semaines |
|
spésolimab |
300 mg par voie sous-cutanée |
150 mg par voie sous-cutanée toutes les 12 semaines |
|
Placebo |
Administration par voie souscutanée |
Administration par voie sous-cutanée toutes les 4 semaines |
Les patients de l'étude étaient à 38,2 % de sexe masculin et à 61,8 % de sexe féminin. L'âge moyen était de 40,4 ans (intervalle : 14 à 75 ans). La population de l'étude comptait 8 patients adolescents (6,5 % ; 2 dans chaque groupe de traitement) et était composée de 64,2 % de patients asiatiques et de 35,8 % de patients caucasiens. Les patients inclus dans l'étude présentaient un sous-score de pustulation de 1 (28,5 %) ou de 0 (71,5 %) à l'échelle GPPGA et un score total de 1 (86,2 %) ou 0 (13,8 %) à l'échelle GPPGA. Au moment de la randomisation, 74,8 % des patients recevaient un traitement systémique du PPG, qui a été arrêté à l'instauration du traitement de l'étude.
Bien que 3 schémas posologiques ont été étudiés dans l'essai Effisayil 2, le schéma posologique de spésolimab recommandé pour la prévention des poussées de PPG est d'une dose de charge de 600 mg par voie sous-cutanée suivie de 300 mg par voie sous-cutanée toutes les 4 semaines (voir rubrique Posologie et mode d'administration). Les résultats présentés ci-dessous se rapportent au schéma posologique recommandé.
En cas de poussée, les patients pouvaient recevoir jusqu'à 2 doses de 900 mg de spésolimab par voie intraveineuse en ouvert (voir rubrique Posologie et mode d'administration). Deux patients du groupe spésolimab à la dose recommandée (6,7 %) et 15 patients du groupe placebo (48,4 %) ont reçu le traitement aigu par voie intraveineuse.
En comparaison au placebo, le traitement par spésolimab à la dose recommandée a montré une amélioration statistiquement significative du critère principal et du principal critère secondaire (voir tableau 5).
Tableau 5 : Délai de survenue de la première poussée de PPG et survenue d'au moins une poussée de PPG jusqu'à la semaine 48 (Effisayil 2)
|
|
Placebo |
|
Spésolimab à la dose recommandée |
|
Nombre de patients analysés, N |
31 |
|
30 |
|
Patients ayant présenté des poussées de PPG, N (%)* |
16 (51,6) |
|
3 (10,0) |
|
Hazard ratio (HR)** pour le délai de survenue de la première poussée, par rapport au placebo (IC à 95 %) |
|
0,16 (0,05 ; 0,54) |
|
|
Valeur p*** |
|
0,0005 |
|
|
Différence de risque de survenue d'une poussée de PPG, par rapport au placebo (IC à 95 %) |
|
-39,0 % (-62,1 ; -15,9) |
|
|
Valeur p**** |
|
0,0013 |
|
* L'administration de spésolimab par voie intraveineuse ou d'un traitement standard prescrit par l'investigateur, destiné à traiter une aggravation du PPG, était envisagée lors de la survenue d'une poussée de PPG. ** Modèle de régression de Cox stratifié sur la prise de médicaments systémiques contre le PPG à la randomisation.
*** Test du log rank stratifié sur la prise de médicaments systémiques contre le PPG à la randomisation, valeur p unilatérale.
**** Test de Cochran-Mantel-Haenszel après imputation multiple, stratifié sur la prise de médicaments systémiques contre le PPG à la randomisation, valeur p unilatérale.
L'efficacité de la dose recommandée de spésolimab par voie sous-cutanée par rapport au placebo a été observée peu de temps après la randomisation et s'est maintenue jusqu'à la semaine 48 (figure 1).
Figure 1 : Délai de survenue de la première poussée de PPG jusqu'à la semaine 48 (Effisayil 2)
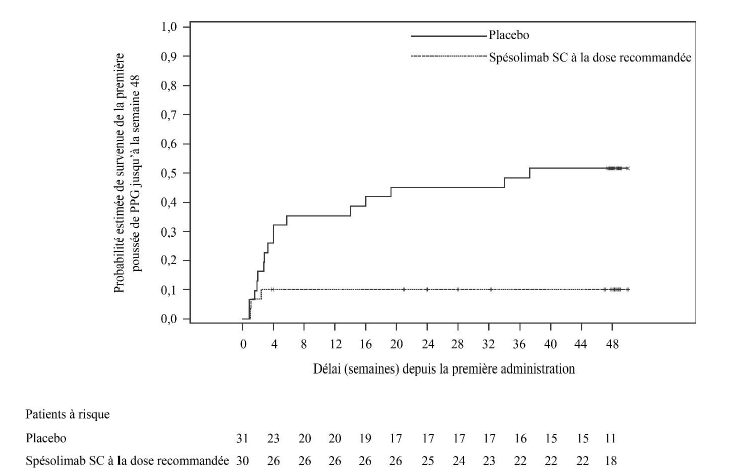
Un effet du traitement a été observé chez tous les patients, quel que soit leur statut mutationnel IL36RN, pour le critère d'évaluation principal et pour le principal critère d'évaluation secondaire.
Un patient adolescent du groupe placebo a reçu un traitement standard prescrit par l'investigateur, destiné à traiter une aggravation du PPG, et qui a été considéré comme une poussée de PPG. Aucun patient adolescent du groupe spésolimab à la dose recommandée n'a présenté de poussée de PPG.
Au regard des scores aux échelles PSS et DLQI, un effet préventif sur l'aggravation du PPG a également été observé, comme indiqué par le hazard ratio de 0,42 (IC à 95 % : 0,20-0,91) pour le score PSS et de 0,26 (IC à 95 % : 0,11-0,62) pour le score DLQI.
Immunogénicité
Chez les patients atteints de PPG traités par spésolimab par voie intraveineuse dans l'essai Effisayil 1,
46 % ont développé des anticorps anti-médicament (AAM). La majorité des patients positifs aux AAM ont également développé des anticorps neutralisants. Dans l'essai Effisayil 2, après plusieurs doses sous-cutanées de spésolimab, 41 % des patients ont développé des AAM. La majorité des patients positifs aux AAM ont également développé des anticorps neutralisants.
La clairance du spésolimab a augmenté avec les titres d'AAM.
Les données relatives à l'administration d'une nouvelle dose chez des patients ayant développé des AAM (n = 4) sont limitées étant donné que la majorité des patients de l'essai Effisayil 1 n'ont pas présenté de nouvelle poussée. À ce jour, on ignore s'il existe une corrélation entre la présence d'AAM anti-spésolimab et le maintien de l'efficacité en cas de traitement d'une poussée. Après administration de spésolimab par voie sous-cutanée dans l'essai Effisayil 2, la présence d'AAM n'a pas eu d'incidence manifeste sur l'efficacité ou la sécurité.
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec Spevigo dans la population pédiatrique de moins de 12 ans dans le traitement du psoriasis pustuleux généralisé (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Autorisation de mise sur le marché conditionnelle
Une autorisation de mise sur le marché « conditionnelle » a été délivrée pour ce médicament. Cela signifie que des preuves supplémentaires concernant ce médicament sont attendues. L'Agence européenne des médicaments réévaluera toute nouvelle information sur ce médicament au moins chaque année et, si nécessaire, ce RCP sera mis à jour.
Un modèle pharmacocinétique de population a été élaboré sur la base des données recueillies chez des volontaires sains, chez des patients atteints de PPG et chez des patients présentant d'autres maladies. Après une dose unique de 900 mg en intraveineuse, l'ASC0-8 (IC à 95 %) et la Cmax (IC à 95 %) estimées par le modèle pharmacocinétique de population chez un patient type atteint de PPG n'ayant pas développé d'anticorps anti-médicament (AAM) étaient respectivement de 4 750 (4 510 ; 4 970) µg·j/mL et de 238 (218 ; 256) µg/mL. Après l'administration d'une dose de charge de 600 mg de spésolimab par voie sous-cutanée suivie d'une dose de 300 mg de spésolimab par voie sous-cutanée toutes les 4 semaines, la concentration résiduelle moyenne (%CV) à l'état d'équilibre était comprise entre 33,4 µg/mL (37,6 %) et 42,3 µg/mL (43,0 %).
Absorption
Après administration sous-cutanée d'une dose unique de spésolimab chez des volontaires sains, les concentrations plasmatiques maximales ont été atteintes au bout de 5,5 à 7,0 jours. Après administration sous-cutanée dans l'abdomen, la biodisponibilité absolue était légèrement plus élevée aux doses supérieures, avec des valeurs estimatives de 58 %, 65 % et 72 % aux doses de 150 mg, 300 mg et 600 mg respectivement. Sur la base de données limitées, la biodisponibilité absolue était d'environ 85 % après administration sous-cutanée de 300 mg de spésolimab dans la cuisse.
Distribution
D'après l'analyse pharmacocinétique de la population, le volume de distribution habituel à l'état d'équilibre était de 6,4 L.
Biotransformation
La voie métabolique du spésolimab n'a pas été caractérisée. S'agissant d'un anticorps monoclonal IgG1 humanisé, il est attendu que le spésolimab se dégrade en petits peptides et acides aminés par catabolisme, d'une manière comparable aux IgG endogènes.
Élimination
Dans l'intervalle de dose linéaire (0,3 à 20 mg/kg), d'après le modèle pharmacocinétique de population, la clairance du spésolimab (IC à 95 %) chez un patient type atteint de PPG n'ayant pas développé d'AAM et pesant 70 kg était de 0,184 L/j. La demi-vie terminale était de 25,5 jours.
Linéarité/non-linéarité
Après administration d'une dose unique par voie intraveineuse, le spésolimab a présenté une pharmacocinétique linéaire avec une augmentation de l'exposition proportionnelle à la dose à des doses comprises entre 0,3 et 20 mg/kg. La clairance (Cl) et la demi-vie terminale étaient toutes deux indépendantes de la dose. Après administration d'une dose unique par voie sous-cutanée, l'exposition au spésolimab a augmenté légèrement plus que proportionnellement à la dose à des doses comprises entre 150 mg et 600 mg, car sa biodisponibilité est légèrement supérieure à des doses plus élevées.
Poids corporel
Les concentrations de spésolimab étaient plus faibles chez les patients de poids corporel plus élevé, et plus élevées chez les patients de poids corporel plus faible. Le spésolimab n'a pas été étudié chez les patients atteints de PPG pesant plus de 164 kg.
D'après les modélisations et simulations pharmacocinétiques, la dose recommandée chez les adolescents âgés de 12 ans ou plus et pesant ≥ 30 et < 40 kg correspond à la moitié de la dose recommandée pour les adultes et les adolescents âgés de 12 ans ou plus et pesant au moins 40 kg (voir rubrique Posologie et mode d'administration).
Chez les patients pesant ≥ 30 et < 40 kg et recevant la posologie réduite, l'exposition devrait être comparable à celle observée lors des études menées dans l'indication de PPG.
Sujets âgés/sexe/origine ethnique
D'après les analyses pharmacocinétiques de population, l'âge, le sexe et l'origine ethnique n'ont aucun effet cliniquement significatif sur la pharmacocinétique du spésolimab.
Insuffisance hépatique et insuffisance rénale
Le spésolimab étant un anticorps monoclonal, il n'est pas attendu qu'il soit éliminé par voie hépatique ou rénale. Aucun essai formel n'a évalué l'effet d'une insuffisance hépatique ou rénale sur la pharmacocinétique du spésolimab.
L'analyse pharmacocinétique de population n'a pas identifié que l'exposition systémique au spésolimab était influencée par une insuffisance hépatique légère ou par une insuffisance rénale légère ou modérée.
Population pédiatrique
La pharmacocinétique du spésolimab n'a pas été étudiée chez les patients pédiatriques âgés de moins de 14 ans.
La pharmacocinétique plasmatique du spésolimab observée chez les adolescents était cohérente avec celle observée chez les adultes.
Spevigo n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Les données non cliniques issues des études de toxicologie en administration répétée n'ont pas révélé de risque particulier pour l'homme.
Toxicité sur le développement et les fonctions de reproduction
Les études non cliniques menées chez la souris avec un substitut d'anticorps monoclonal anti-IL36R murin n'ont pas mis en évidence d'effets délétères directs ou indirects sur la gestation, le développement embryofœtal ou la fertilité.
Génotoxicité
Aucune étude de génotoxicité n'a été réalisée avec le spésolimab.
Cancérogénèse
Aucune étude de cancérogenèse ou de mutagenèse n'a été réalisée avec le spésolimab.
Ce médicament est compatible avec les kits de perfusion composés de polychlorure de vinyle (PVC), de polyéthylène (PE), de polypropylène (PP), de polybutadiène ou de polyuréthane (PUR), ainsi qu'avec les filtres à membrane intégrés composés de polyéthersulfone (PES, neutre ou chargé positivement) ou de polyamide (PA) chargé positivement.
Instructions de manipulation
- Procéder à une inspection visuelle du flacon avant utilisation. Le flacon doit être éliminé si la solution est trouble, a changé de couleur ou contient des particules colorées ou de grande taille.
- Spevigo est réservé à un usage unique.
- Une technique aseptique doit être utilisée pour préparer la solution pour perfusion :
o Pour la dose recommandée de 900 mg, retirer et éliminer 15 mL d'un récipient contenant 100 mL de solution injectable de chlorure de sodium à 9 mg/mL (0,9 %), et à la place injecter lentement 15 mL de solution à diluer stérile de spésolimab (deux flacons de 450 mg/7,5 mL). o Pour la dose recommandée de 450 mg, retirer et éliminer 7,5 mL d'un récipient contenant 100 mL de solution injectable de chlorure de sodium à 9 mg/mL (0,9 %), et à la place injecter lentement 7,5 mL de solution à diluer stérile de spésolimab (un flacon de 450 mg/7,5 mL).
o Mélanger doucement avant utilisation. La solution pour perfusion de spésolimab diluée doit être utilisée immédiatement.
- Spevigo ne doit pas être mélangé avec d'autres médicaments. Un accès veineux préexistant peut être utilisé pour administrer la solution pour perfusion de spésolimab diluée, dès lors que les informations de compatibilité précédemment décrites sont prises en compte. La tubulure doit être rincée avec une solution injectable de chlorure de sodium à 9 mg/mL (0,9 %) avant et après la perfusion. Aucune autre perfusion ne doit être administrée en parallèle par le même accès veineux.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I
Prescription réservée aux spécialistes et services DERMATOLOGIE
Prescription réservée aux spécialistes et services PEDIATRIE
Réservé à l'usage HOSPITALIER
Solution à diluer pour perfusion (solution à diluer stérile)
Solution limpide à légèrement opalescente, incolore à légèrement jaune-brun.
Flacon en verre (de type I) incolore de 10 mL muni d'un bouchon en caoutchouc enduit et d'un opercule serti en aluminium avec un bouton en plastique bleu, contenant 7,5 mL de solution à diluer.
Présentation : 2 flacons.
Chaque flacon contient 450 mg de spésolimab dans 7,5 mL.
Chaque mL de solution à diluer pour perfusion contient 60 mg de spésolimab.
Après dilution, chaque mL de solution contient 9 mg de spésolimab (voir la rubrique Précautions particulières d'élimination et de manipulation).
Le spésolimab est produit à partir de cellules ovariennes de hamster chinois par la technologie de l'ADN recombinant.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Acétate de sodium trihydraté (E262)
Acide acétique glacial (E260) (pour l'ajustement du pH)
Saccharose
Chlorhydrate d'arginine
Polysorbate 20 (E432)
Eau pour préparations injectables