
EVRENZO 150 mg, comprimé pelliculé, boîte de 12 plaquettes thermoformées de 1
Dernière révision : 30/09/2022
Taux de TVA : 2.1%
Prix de vente : 237,67 €
Taux remboursement SS : 65%
Base remboursement SS : 237,67 €
Laboratoire exploitant : ASTELLAS PHARMA FRANCE
Source :

Evrenzo est indiqué dans le traitement des patients adultes présentant une anémie symptomatique associée à une maladie rénale chronique (MRC).
Evrenzo est contre-indiqué dans les situations suivantes :
- Hypersensibilité à la substance active, à l‘arachide, au soja ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
- Troisième trimestre de la grossesse (voir rubriques Mises en garde spéciales et précautions d'emploi et Fertilité, grossesse et allaitement).
-
Allaitement (voir rubrique Fertilité, grossesse et allaitement).
Risque cardiovasculaire et de mortalité
Globalement, en se basant sur les données de comparaison directe des
traitements par roxadustat et ASE, le risque cardiovasculaire et de
mortalité associé au traitement par roxadustat a été estimé comparable
au risque cardiovasculaire et de mortalité du traitement par ASE (voir
rubrique Propriétés pharmacodynamiques). Dans la mesure où,
pour les patients non dialysés présentant une anémie associée à une
MRC, ce risque n'a pas pu être estimé avec une confiance suffisante par
rapport au placebo, la décision de traiter ces patients par roxadustat
doit être basée sur des critères similaires à ceux qui seraient
appliqués avant un traitement par ASE. De plus, plusieurs facteurs
participant à ce risque ont été déterminés, notamment l'absence de
réponse au traitement et le changement de traitement chez les patients
dialysés stables sous traitement par un ASE (voir rubriques Posologie et mode d'administration et Propriétés pharmacodynamiques). En cas d'absence de réponse, le traitement par roxadustat ne doit pas être poursuivi après la 24ème semaine de traitement (voir rubrique Posologie et mode d'administration).
Le changement de traitement des patients dialysés stables sous
traitement par un ASE ne doit être envisagé que s'il existe une raison
clinique valable (voir rubrique Posologie et mode d'administration).
Chez les patients non dialysés stables sous un traitement par ASE et
présentant une anémie associée à une MRC, ce risque n'a pas pu être
évalué puisque cette population de patients n'a pas été étudiée. La
décision de traiter ces patients par roxadustat doit être basée sur
l'évaluation du rapport bénéfice-risque pour chaque patient.
Évènements vasculaires thrombotiques
Le risque rapporté d'évènements vasculaires thrombotiques (EVT) doit
être soigneusement mis en balance avec les bénéfices retirés du
traitement par roxadustat, en particulier chez les patients ayant des
facteurs de risque préexistants d'EVT, y compris une obésité et un
antécédent d'EVT (ex. thrombose veineuse profonde [TVP] et embolie
pulmonaire [EP]). La thrombose veineuse profonde a été rapportée comme
fréquente et l'embolie pulmonaire comme peu fréquente chez les patients
ayant participé aux études cliniques. La majorité des évènements de
type TVP et EP était grave.
Lors des études cliniques, la thrombose de l'accès vasculaire (TAV) a été rapportée comme très fréquente parmi les patients atteints d'une MRC dialysés (voir rubrique Effets indésirables).
Chez les patients atteints d'une MRC dialysés, les taux de TAV chez les patients traités par roxadustat étaient plus élevés dans les 12 premières semaines après l'instauration du traitement, à des taux d'Hb supérieurs à 12 g/dL et dont l'Hb a augmenté de plus de 2 g/dL en 4 semaines. Il est recommandé de surveiller le taux d'Hb et d'adapter la dose en suivant les règles d'adaptation posologique (voir tableau 2), afin d'éviter que ce taux soit supérieur à 12 g/dL et augmente de plus de 2 g/dL sur4 semaines.
Les patients présentant des signes et des symptômes d'EVT doivent être rapidement évalués et traités suivant le traitement de référence. La décision d'interrompre ou d'arrêter définitivement le traitement doit être basée sur l'évaluation du rapport bénéfice-risque pour chaque patient.
Convulsions
Les convulsions ont également été rapportées comme fréquentes parmi les
patients inclus dans les études cliniques ayant reçu du roxadustat
(voir rubrique Effets indésirables). Le roxadustat doit être
utilisé avec précaution chez les patients ayant des antécédents
d'épilepsie (convulsions ou crises d'épilepsie), des épilepsies ou des
affections médicales associées à une prédisposition à une activité
convulsive telle que les infections du système nerveux central (SNC).
La décision d'interrompre ou d'arrêter définitivement le traitement
doit être basée sur l'évaluation du rapport bénéfice-risque pour chaque
patient.
Infections graves
Les infections graves les plus fréquemment rapportées ont été la
pneumonie et les infections des voies urinaires. Les patients
présentant des signes et des symptômes d'infection doivent être
rapidement évalués et traités suivant le traitement de référence.
Sepsis
Le sepsis était l'une des infections graves les plus fréquemment
rapportées et incluait des évènements fatals. Les patients présentant
des signes et des symptômes de sepsis (ex. une infection qui se propage
dans tout le corps et s'accompagne d'une pression artérielle basse et
d'un risque de défaillance d'organe) doivent être rapidement évalués et
traités suivant le traitement de référence.
Hypothyroïdie secondaire
Des cas d'hypothyroïdie secondaire ont été rapportés lors du traitement par roxadustat (voir rubrique Effets indésirables).
Ces réactions étaient réversibles à l'arrêt du roxadustat. La
surveillance de la fonction thyroïdienne est recommandée si elle est
cliniquement indiquée.
Réponse inadéquate au traitement
Une réponse inadéquate au traitement par roxadustat doit conduire
rapidement à en rechercher les causes. Les carences nutritionnelles
doivent être corrigées. Des infections intercurrentes, une perte de
sang occulte, une hémolyse, une intoxication sévère par l'aluminium,
des maladies hématologiques sous-jacentes ou une myélofibrose peuvent
aussi compromettre la réponse érythropoïétique. La numération des
réticulocytes doit être envisagée comme élément de l'évaluation. Si les
causes habituelles de non-réponse ont été exclues et si le patient
présente une réticulocytopénie, un examen de la moelle osseuse doit
être envisagé. Le traitement par Evrenzo ne doit pas être poursuivi
au-delà de 24 semaines en cas de réponse inadéquate pour laquelle une
cause résoluble n'a pas été trouvée.
Insuffisance hépatique
La prudence est de mise lorsque le roxadustat est administré à des
patients présentant une insuffisance hépatique modérée (Child-Pugh
classe B). L'utilisation d'Evrenzo n'est pas recommandée chez les
patients présentant une insuffisance hépatique sévère (Child-Pugh
classe C) (voir rubrique Propriétés pharmacocinétiques).
Grossesse et contraception
Le roxadustat ne doit pas être instauré chez les femmes qui prévoient
une grossesse, pendant la grossesse ou lorsqu'une anémie associée à la
MRC est diagnostiquée pendant la grossesse. Dans ces cas, un autre
traitement doit être débuté, le cas échéant. Si une grossesse débute au
cours du traitement par roxadustat, celui-ci doit être interrompu et un
autre traitement instauré, le cas échéant. Les femmes en âge de
procréer doivent utiliser une contraception hautement efficace pendant
le traitement et pendant une semaine au minimum après la dernière dose
d'Evrenzo (voir rubriques Contre-indications et Fertilité, grossesse et allaitement).
Mésusage
Un mésusage peut conduire à une augmentation excessive de
l'hématocrite, qui peut être associée à des complications
cardiovasculaires pouvant engager le pronostic vital.
Excipients
Evrenzo contient du lactose. Les patients présentant des problèmes
héréditaires rares d'intolérance au galactose, de déficit total en
lactase ou de malabsorption du glucose et du galactose ne doivent pas
prendre ce médicament.
Evrenzo contient de la laque d'aluminium rouge Allura AC (voir rubrique Liste des excipients) qui peut provoquer des réactions allergiques.
Evrenzo contient des traces de lécithine de soja. Les patients qui sont
allergiques à l'arachide ou au soja, ne doivent pas utiliser ce
médicament.
Résumé du profil de sécurité
La sécurité d'Evrenzo a été évaluée chez 3 542 patients non dépendants de la dialyse (NDD) et 3 353 patients dépendants de la dialyse (DD) présentant une anémie et une MRC, qui ont reçu au moins une dose de roxadustat.
Les effets indésirables les plus fréquents (≥ 10 %) associés au roxadustat sont l'hypertension (13,9 %), la thrombose de l'accès vasculaire (12,8 %), la diarrhée (11,8 %), l'œdème périphérique (11,7 %), l'hyperkaliémie (10,9 %) et la nausée (10,2 %).
Les effets indésirables graves les plus fréquents (≥ 1 %) associés au roxadustat ont été le sepsis (3,4 %), l'hyperkaliémie (2,5 %), l'hypertension (1,4 %) et la thrombose veineuse profonde (1,2 %).
Liste tabulée des effets indésirables
Les effets indésirables observés au cours des études cliniques et/ou de
l'expérience post- commercialisation sont répertoriés dans cette
rubrique par catégorie de fréquence.
Les catégories de fréquence sont définies comme suit : très fréquent (≥
1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, <
1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000) ;
fréquence indéterminée (ne peut être estimée sur la base des données
disponibles).
Tableau 3. Effets indésirables
| Classe de systèmes d'organes MedDRA (SOC) | Catégorie de fréquence | Effet indésirable |
| Infections et infestations | Fréquent | Sepsis |
| Affections endocriniennes | Fréquence indéterminée | Hypothyroïdie secondaire |
| Troubles du métabolisme et de la nutrition | Très fréquent | Hyperkaliémie |
| Affections psychiatriques | Fréquent | Insomnie |
| Affections du système nerveux | Fréquent | Convulsions, céphalée |
| Affections vasculaires | Très fréquent | Hypertension, thrombose de l'accès vasculaire (TAV)1 |
| Fréquent | Thrombose veineuse profonde (TVP) | |
| Affections de la peau et du tissu sous-cutané | Fréquence indéterminée | Dermatite exfoliative généralisée (DEG) |
| Affections gastro-intestinales | Très fréquent | Nausée, diarrhée |
| Fréquent | Constipation, vomissement | |
| Affections hépatobiliaires | Peu fréquent | Hyperbilirubinémie |
| Affections respiratoires, thoraciques et médiastinales | Peu fréquent | Embolie pulmonaire |
| Troubles généraux et anomalies au site d'administration | Très fréquent | Œdème périphérique |
| Investigations | Fréquence indéterminée | Diminution des taux sanguins de thyréostimuline (TSH) |
Description de certains effets indésirables
Évènements vasculaires thrombotiques
Chez les patients atteints d'une MRC NDD, les événements de TVP ont été
peu fréquents et sont survenus chez 1,0 % (0,6 patient avec évènements
sur 100 patient-années d'exposition) dans le groupe roxadustat, et chez
0,2 % (0,2 patient avec évènements sur 100 patient-années d'exposition)
dans le groupe placebo. Chez les patients atteints d'une MRC DD, les
évènements de TVP sont survenus chez 1,3 % (0,8 patient avec évènements
sur 100 patient-années d'exposition) dans le groupe roxadustat, et chez
0,3 % (0,1 patient avec évènements sur 100 patient-années d'exposition)
dans le groupe ASE (voir rubrique Mises en garde spéciales et précautions d'emploi).
Chez les patients atteints d'une MRC NDD, une embolie pulmonaire a été observée chez 0,4 % (0,2 patient avec évènements sur 100 patient-années d'exposition) dans le groupe roxadustat, comparativement à 0,2 % (0,1 patient avec évènements sur 100 patient-années d'exposition) dans le groupe placebo. Chez les patients atteints d'une MRC DD, une embolie pulmonaire a été observée chez 0,6 % (0,3 patient avec évènements sur 100 patient-années d'exposition) dans le groupe roxadustat, comparativement à 0,5 % (0,3 patient avec évènements sur 100 patient-années d'exposition) dans le groupe ASE (voir rubrique Mises en garde spéciales et précautions d'emploi).
Chez les patients atteints d'une MRC DD, une thrombose de l'accès vasculaire a été observée chez 12,8 % (7,6 patients avec évènements sur 100 patient-années d'exposition) dans le groupe roxadustat, comparativement à 10,2 % (5,4 patients avec évènements sur 100 patient-années d'exposition) dans le groupe ASE (voir rubrique Mises en garde spéciales et précautions d'emploi).
Convulsions
Chez les patients atteints d'une MRC NDD, des convulsions sont
survenues chez 1,1 % (0,6 patient avec évènements sur 100
patient-années d'exposition) dans le groupe roxadustat, et chez 0,2 %
(0,2 patient avec évènements sur 100 patient-années d'exposition) dans
le groupe placebo (voir rubrique Mises en garde spéciales et précautions d'emploi).
Chez les patients atteints d'une MRC DD, des convulsions sont survenues chez 2,0 % (1,2 patient avec évènements sur 100 patient-années d'exposition) dans le groupe roxadustat, et chez 1,6 % (0,8 patient avec évènements sur 100 patient-années d'exposition) dans le groupe ASE (voir rubrique Mises en garde spéciales et précautions d'emploi).
Sepsis
Chez les patients atteints d'une MRC NDD, un sepsis a été observé chez
2,1 % (1,3 patient avec évènements sur 100 patient-années d'exposition)
dans le groupe roxadustat, comparativement à 0,4 % (0,3 patient avec
évènements sur 100 patient-années d'exposition) dans le groupe placebo.
Chez les patients dialysés, un sepsis a été observé chez 3,4 % (2,0
patients avec évènements sur 100 patient- années d'exposition) dans le
groupe roxadustat, comparativement à 3,4 % (1,8 patient avec évènements
sur 100 patient-années d'exposition) dans le groupe ASE (voir rubrique Mises en garde spéciales et précautions d'emploi).
Réactions cutanées
La dermatite exfoliative généralisée, faisant partie des effets
indésirables cutanés sévères (SCAR), a été rapportée au cours de la
surveillance post-commercialisation et a montré un lien avec le
traitement par roxadustat (fréquence inconnue).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du
médicament est importante. Elle permet une surveillance continue du
rapport bénéfice/risque du médicament. Les professionnels de santé
déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
Le
changement de traitement des patients non dépendants de la dialyse et
stables sous traitement par un ASE n'a pas été étudié. La décision de
traiter ces patients par roxadustat doit être basée sur l'évaluation du
rapport bénéfice-risque pour chaque patient.
Le changement de traitement des patients dialysés stables sous
traitement par un ASE ne doit être envisagé que s'il existe une raison
clinique valable.
AVANT INSTAURATION DU TRAITEMENT ;
- Evaluer toutes les autres causes d'anémie.
- S'assurer que les réserves en fer sont suffisantes.
Surveillance du traitement :
- CONTROLER les tauxd'Hb toutes les deux semaines jusqu'à l'obtention
du taux cible compris entre 10 et 12 g/dL et sa stabilisation, puis
toutes les 4 semaines, ou selon la pertinence clinique.
NE PAS poursuivre le traitement par roxadustat au-delà de 24 semaines
si une augmentation cliniquement significative des taux d'Hb n'est pas
atteinte. Rechercher alors d'autres causes d'une réponse inadéquate et
les traiter avant de reprendre le traitement par roxadustat.
- Surveillance de la fonction thyroïdienne recommandée si cliniquement indiquée.
CONTACTER IMMEDIATEMENT LE MEDECIN en cas de :
- Douleur et/ou gonflement des jambes, crampes ou sensation de chaleur dans une jambe.
- Difficulté respiratoire soudaine, douleur dans la poitrine, sensation
d'anxiété, de vertige, de tête vide ou évanouissement, accélération du
coeur, toux (parfois avec du sang).
- Caillot au niveau du point d'accès pour l'hémodialyse pouvant
provoquer un gonflement, une rougeur, un durcissement ou un
épaississement de la peau autour de l'accès, un suintement au niveau du
site d'accès ou l'absence de vibration ressentie au niveau de la zone
d'accès.
- Epilepsie (convulsion ou crise d'épilepsie) ou d'éventuels signes
avant-coureurs d'une crise d'épilepsie (mal de tête, irritabilité,
peur, confusion ou sensations inhabituelles).
- Fièvre, sueurs ou frissons, mal de gorge, nez qui coule,
essoufflement, sensation de faiblesse ou d'évanouissement, confusion,
toux, vomissements, diarrhée ou douleur à l'estomac, brûlures lors de
la miction, peau rouge ou douloureuse : tout signe traduisant une
infection.
PRUDENCE en cas de conduite de véhicules ou d'utilisation de machines (convulsions possibles).
Femme en âge de procréer : utiliser une contraception hautement
efficace pendant le traitement et pendant une semaine au minimum après
la dernière dose.
Grossesse, femmes en âge de procréer et contraception
Il n'existe pas de données sur l'utilisation du roxadustat chez la
femme enceinte. Les études effectuées chez l'animal ont montré une
toxicité sur la reproduction (voir rubrique Données de sécurité préclinique).
Le roxadustat est contre-indiqué pendant le troisième trimestre de grossesse (voir rubriques Contre-indications et Mises en garde spéciales et précautions d'emploi.).
Le roxadustat n'est pas recommandé pendant le premier et le deuxième trimestre de grossesse (voir rubrique Mises en garde spéciales et précautions d'emploi.).
Si une grossesse débute au cours du traitement avec Evrenzo, celui-ci doit être interrompu et d'autres traitements instaurés, le cas échéant (voir rubrique Contre-indications).
Allaitement
On ne sait pas si le roxadustat/ses métabolites sont excrétés dans le
lait maternel. Les données disponibles chez l'animal ont montré
l'excrétion du roxadustat dans le lait (voir rubrique Données de sécurité préclinique). Evrenzo est contre-indiqué pendant l'allaitement (voir rubriques Contre-indications et Données de sécurité préclinique).
Fertilité
Dans les études conduites chez l'animal, aucun effet du roxadustat sur
la fertilité des mâles et des femelles n'a été observé. Toutefois, des
modifications des organes reproducteurs du rat mâle ont été observées.
Les effets potentiels du roxadustat sur la fertilité masculine humaine
sont actuellement inconnus. À une dose maternellement toxique, une
perte embryonnaire augmentée a été observée (voir rubrique Données de sécurité préclinique).
Les femmes en âge de procréer doivent utiliser une contraception
hautement efficace pendant le traitement et pendant une semaine au
minimum après la dernière dose d'Evrenzo.
Effet des médicaments sur le roxadustat
Chélateurs du phosphate et autres produits contenant des cations multivalents
La coadministration de roxadustat et de chélateurs du phosphate tels
que le carbonate de sévélamer ou l'acétate de calcium chez des sujets
sains a diminué l'ASC du roxadustat de 67 % et 46 % et la Cmax
de 66 % et 52 %, respectivement. Le roxadustat peut former un chélate
avec des cations multivalents tels que dans les chélateurs du phosphate
ou d'autres produits contenant du calcium, du fer, du magnésium ou de
l'aluminium. L'administration décalée dans le temps de chélateurs du
phosphate (en respectant un intervalle d'au moins une heure) n'a eu
aucun effet clinique significatif sur l'exposition au roxadustat chez
les patients atteints d'une MRC. Le roxadustat doit être pris au moins
1 heure après l'administration de chélateurs du phosphate ou d'autres
médicaments ou compléments contenant des cations multivalents (voir
rubrique Posologie et mode d'administration). Cette restriction
ne s'applique pas au carbonate de lanthane, car la coadministration de
roxadustat et de carbonate de lanthane n'a pas conduit à un changement
cliniquement significatif de l'exposition plasmatique du roxadustat.
Modificateurs de l'activité du CYP2C8 ou de l'UGT1A9
Le roxadustat est un substrat du CYP2C8 et de l'UGT1A9. La
coadministration de roxadustat et de gemfibrozil (inhibiteur du CYP2C8
et de l'OATP1B1) ou de probénécide (inhibiteur de l'UGT et de
l'OAT1/OAT3) chez des sujets sains a augmenté l'ASC du roxadustat de
2,3 fois et la Cmax de 1,4
fois. Les taux d'Hb doivent être surveillés lors de l'instauration ou
de l'interruption d'un traitement concomitant par gemfibrozil,
probénécide, d'autres puissants inhibiteurs ou inducteurs du CYP2C8, ou
puissants inhibiteurs de l'UGT1A9. Adapter la dose de roxadustat en
suivant les règles d'adaptation posologique (voir tableau 2) en
fonction de la surveillance des taux d'Hb.
Effet du roxadustat sur d'autres médicaments
Substrats de l'OATP1B1 ou de la BCRP
Le roxadustat est un inhibiteur de la BCRP et de l'OATP1B1. Ces
transporteurs jouent un rôle important dans l'assimilation intestinale
et hépatique et l'efflux de statines. La coadministration de 200 mg de
roxadustat et de simvastatine chez des sujets sains a augmenté l'ASC et
la Cmax de la simvastatine de 1,8 et 1,9 fois, respectivement, et l'ASC et la Cmax
de la simvastatine acide (le métabolite actif de la simvastatine) de
1,9 et 2,8 fois, respectivement. Les concentrations de simvastatine et
de simvastatine acide ont également augmenté lorsque la simvastatine
était administrée 2 heures avant ou 4 ou 10 heures après le roxadustat.
La coadministration de 200 mg de roxadustat et de rosuvastatine a
augmenté l'ASC et la Cmax de
la rosuvastatine de 2,9 et 4,5 fois, respectivement. La
coadministration de 200 mg de roxadustat et d'atorvastatine a augmenté
l'ASC et la Cmax de l'atorvastatine de 2,0 et 1,3 fois, respectivement.
Des interactions sont également susceptibles de se produire avec d'autres statines. Lorsqu'une statine est co-administrée avec le roxadustat, tenir compte de cette interaction, surveiller la survenue d'effets indésirables associés aux statines et évaluer la nécessité d'une réduction de la dose de statine. Se reporter aux informations de prescription lors de la prise de décision concernant la dose de statine appropriée pour le patient concerné.
Le roxadustat peut augmenter l'exposition plasmatique d'autres médicaments qui sont des substrats de la BCRP ou de l'OATP1B1. Il convient de surveiller les possibles effets indésirables des médicaments co-administrés et d'adapter la dose en conséquence.
Roxadustat et les ASE
Il est déconseillé de combiner l'administration de roxadustat et d'ASE car cette association n'a pas été étudiée.
Le traitement par roxadustat doit être instauré par un médecin expérimenté dans la prise en charge de l'anémie. Toutes les autres causes d'anémie doivent être évaluées avant le début du traitement par Evrenzo et lors de la décision d'augmenter la dose.
Les symptômes et les séquelles de l'anémie peuvent varier en fonction de l'âge, du sexe et du fardeau de la maladie ; l'évaluation par un médecin de l'évolution et de l'état cliniques du patient est nécessaire. Outre la présence de symptômes d'anémie, des critères tels que la rapidité de la chute de la concentration en hémoglobine (Hb), la réponse antérieure à un traitement à base de fer et le risque de devoir recourir à une transfusion de globules rouges (GR), peuvent avoir un intérêt dans l'évaluation de l'évolution et de l'état cliniques du patient.
Posologie
La dose adéquate de roxadustat doit être prise par voie orale trois fois par semaine lors de jours non- consécutifs.
La dose doit être individualisée afin de parvenir à des taux d'Hb cibles de 10 à 12 g/dL et de les maintenir, comme décrit ci-dessous.
Le traitement par roxadustat ne doit pas être poursuivi au-delà de 24 semaines si une augmentation cliniquement significative des taux d'Hb n'est pas atteinte. Il convient alors de rechercher d'autres causes d'une réponse inadéquate et de les traiter avant de reprendre Evrenzo.
Dose initiale lors de l'instauration du traitement
Il convient de s'assurer que les réserves en fer sont suffisantes avant d'instaurer le traitement.
Patients n'étant pas en cours de traitement par un agent stimulant l'érythropoïèse (ASE)
Pour les patients qui débutent le traitement de leur anémie et qui
n'ont pas été traités auparavant par un ASE, la dose initiale
recommandée de roxadustat est de 70 mg trois fois par semaine chez les
patients pesant moins de 100 kg et de 100 mg trois fois par semaine
chez les patients pesant 100 kg et plus.
Patients en cours de traitement par un ASE
Le traitement des patients actuellement sous ASE peut être changé par
un traitement par roxadustat. Toutefois, le changement de traitement
des patients dialysés stables sous traitement par un ASE ne doit être
envisagé que s'il existe une raison clinique valable (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacodynamiques).
Le changement de traitement des patients non dépendants de la dialyse et stables sous traitement par un ASE n'a pas été étudié. La décision de traiter ces patients par roxadustat doit être basée sur l'évaluation du rapport bénéfice-risque pour chaque patient.
La dose initiale recommandée de roxadustat est basée sur la dose moyenne d'ASE prescrite au cours des 4 semaines précédant ce changement (voir le tableau 1).
La première dose de roxadustat doit remplacer la prochaine dose programmée de l'ASE actuel.
Table 1. Doses initiales de roxadustat à prendre trois fois par semaine suivant la dose d'ASE sous laquelle était le patient
| Dose de darbépoétine alfa par voie intraveineuse ou sous-cutanée (microgrammes/semaine) | Dose d'époétine par voie intraveineuse ou sous- cutanée (UI/semaine) | Dose de méthoxy polyéthylène glycol- époétine bêta par voie intraveineuse ou sous- cutanée (microgrammes/mois) | Dose de roxadustat (milligrammes trois fois par semaine) |
| Inférieure à 25 | Inférieure à 5 000 | Inférieure à 80 | 70 |
| De 25 à inférieure à 40 | De 5 000 à 8 000 | De 80 à 120 inclus | 100 |
| De 40 à 80 inclus | Supérieure à 8 000 et jusqu'à 16 000 inclus | Supérieure à 120 et jusqu'à 200 inclus | 150 |
| Supérieure à 80 | Supérieure à 16 000 | Supérieure à 200 | 200 |
Adaptation posologique et surveillance du taux d'Hb
La dose d'entretien individualisée est comprise entre 20 mg et 400 mg trois fois par semaine (voir la rubrique dose maximale recommandée).
Les taux d'Hb doivent être contrôlés toutes les deux semaines jusqu'à
l'obtention du taux cible compris entre 10 et 12 g/dL et sa
stabilisation, puis toutes les4 semaines, ou selon la pertinence clinique.
La dose de roxadustat peut être augmentée ou diminuée progressivement (par paliers) à partir de la dose initiale, 4 semaines après le début du traitement, puis toutes les 4 semaines, sauf si le taux d'Hb augmente de plus de 2 g/dL, auquel cas la dose doit être immédiatement diminuée d'un palier. Lors de l'adaptation de la posologie de roxadustat, il convient de tenir compte du taux d'Hb actuel et de la variation de ce taux au cours des 4 dernières semaines, et de suivre les paliers d'adaptation posologique conformément à l'algorithme décrit dans le tableau 2.
Les adaptations de la posologie par paliers doivent suivre la séquence des doses disponibles :
20 mg - 40 mg - 50 mg - 70 mg - 100 mg - 150 mg - 200 mg - 250 mg - 300 mg - 400 mg (ce dernier palier (400 mg) n'est valable que pour les patients dialysés)
Tableau 2. Règles d'adaptation posologique
| Variation du taux d'Hb au cours des 4 semaines précédentes* | Taux d'Hb actuel (g/dL) : | |||
| Inférieur à 10,5 | De 10,5 à 11,9 | De 12,0 à 12,9 | 13,0 ou supérieur | |
| Variation d'une valeur supérieure à +1,0 g/dL | Pas de changement | Réduire la dose d'un palier | Réduire la dose d'un palier | Suspendre la prise, surveiller le taux d'Hb. Lorsque le taux d'Hb est inférieur à 12,0 g/dL reprendre |
| Variation d'une valeur comprise entre -1,0 et +1,0 g/dL | Augmenter la dose d'un palier | Pas de changement | Réduire la dose d'un palier | |
| Variation d'une valeur | Augmenter la | Augmenter la | Pas de | la prise de |
| inférieure à -1,0 g/dL | dose d'un | dose d'un | changement | roxadustat à une |
| | palier | palier | | dose réduite de |
| | | | | deux paliers |
*Variation du taux d'hémoglobine (Hb) sur la période précédente de 4 semaines = (valeur Hb actuelle) - (valeur Hb précédente obtenue 4 semaines auparavant).
S'il est nécessaire de diminuer la dose d'un patient recevant déjà la dose la plus faible (20 mg trois fois par semaine), ne pas réduire la dose de 20 mg en cassant le comprimé, mais diminuer la fréquence des prises à deux fois par semaine. Si une nouvelle réduction de la dose est requise, la fréquence des prises peut être à nouveau réduite à une fois par semaine.
Dose d'entretien
Après stabilisation du taux d'Hb dans la cible entre 10 et 12 g/dL, la
surveillance des taux d'Hb doit être poursuivie régulièrement et les
règles d'adaptation posologique suivies (voir tableau 2).
Patients débutant une dialyse pendant le traitement par roxadustat
Aucune adaptation posologique particulière n'est nécessaire pour les
patients atteints d'une MRC qui débutent une dialyse pendant leur
traitement par roxadustat. Les règles normales d'adaptation posologique
(voir tableau 2) doivent être suivies.
Traitement concomitant par roxadustat et par inducteurs ou inhibiteurs enzymatique
Lors de l'instauration ou de l'interruption d'un traitement concomitant
par de puissants inhibiteurs (ex. gemfibrozil) ou inducteurs (ex.
rifampicine) du CYP2C8, ou inhibiteurs de l'UGT1A9 (ex. probénécide) :
les taux d'Hb doivent être surveillés régulièrement et les règles
d'adaptation posologique suivies (voir tableau 2 ; voir également les
rubriques Interactions avec d'autres médicaments et autres formes d'interactions et Propriétés pharmacocinétiques).
Dose maximale recommandée
Patients non dialysés ne pas dépasser une dose de roxadustat de 3 mg/kg ou de 300 mg trois fois par semaine, la dose la plus faible étant retenue.
Patients dialysés ne pas dépasser une dose de roxadustat de 3 mg/kg ou de 400 mg trois fois par semaine, la dose la plus faible étant retenue.
Omission de prise
Si une dose est oubliée et si l'intervalle restant avant la dose
suivante prévue est supérieur à 24 heures, la dose omise doit être
prise dès que possible.
Si l'intervalle restant avant la dose suivante prévue est inférieur ou
égal à 24 heures, la dose oubliée ne doit pas être prise et la dose
suivante doit être prise le jour prévu. Dans chaque cas, le calendrier
normal des prises doit reprendre par la suite.
Populations particulières
Personne âgée
Aucune adaptation de la posologie initiale n'est nécessaire chez les patients âgés (voir rubrique Propriétés pharmacocinétiques).
Patients présentant une insuffisance hépatique
En cas d'insuffisance hépatique légère (Child-Pugh classe A), aucune
adaptation de la posologie initiale n'est nécessaire (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacocinétiques).
En cas d'insuffisance hépatique modérée, la prudence est de mise. La dose initiale doit être réduite de moitié ou arrondie à la dose la plus proche de la moitié lors de l'instauration du traitement chez des patients présentant une insuffisance hépatique modérée (Child-Pugh classe B).
En cas d'insuffisance hépatique sévère (ChildPugh- classe C), l'utilisation d'Evrenzo n'est pas recommandée car la sécurité et l'efficacité n'ont pas été évaluées dans cette population (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l'efficacité du roxadustat chez les patients âgés de
moins de 18 ans n'ont pas été établies. Aucune donnée n'est disponible.
Mode d'administration
Les comprimés pelliculés d'Evrenzo doivent être pris par voie orale
avec ou sans nourriture. Les comprimés doivent être avalés entiers et
non mâchés, cassés ou écrasés, en l'absence de données cliniques dans
ces conditions, et afin de protéger le noyau photosensible du comprimé
d'une photodégradation.
Les comprimés doivent être pris au moins 1 heure après l'administration de chélateurs du phosphate (à l'exception du lanthane) ou d'autres médicaments contenant des cations multivalents tels que le calcium, le fer, le magnésium ou l'aluminium (voir rubriques Interactions avec d'autres médicaments et autres formes d'interactions et Propriétés pharmacocinétiques).
Durée de conservation :
4 ans.
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
Des doses uniques suprathérapeutiques de roxadustat 5 mg/kg (jusqu'à 510 mg) administrées à des sujets sains ont été associées à une augmentation transitoire de la fréquence cardiaque, à une fréquence augmentée de douleur musculosquelettique légère à modérée, de céphalées, de tachycardie sinusale et moins fréquemment, à une pression artérielle basse, toutes ces manifestations ayant été non graves. Un surdosage du roxadustat peut élever les taux d'Hb au-dessus du taux cible (10-12 g/dL) ; il nécessited'être pris en charge par un arrêt du traitement par roxadustat ou une réduction de sa posologie (voir rubrique Posologie et mode d'administration) et par une étroite surveillance associée à un traitement symptomatique, selon l'indication clinique. Le roxadustat et ses métabolites ne sont pas significativement éliminés par hémodialyse (voir rubrique Propriétés pharmacocinétiques).
Classe pharmacothérapeutique : préparations antianémiques, autres préparations antianémiques. code ATC : B03XA05.
Mécanisme
d'action
Le roxadustat est un inhibiteur de la prolyl
hydroxylase du facteur induit par l'hypoxie (HIF-PHI). L'activité de l'enzyme
HIF-PH contrôle les taux intracellulaires de HIF, un facteur de transcription
qui régule l'expression des gènes impliqués dans l'érythropoïèse. L'activation
de la voie du HIF est importante dans la réponse adaptative à l'hypoxie pour
augmenter la production d'hématies. Par inhibition réversible de l'enzyme
HIF-PH, le roxadustat stimule une réponse érythropoïétique coordonnée qui inclut l'augmentation des
taux plasmatiques d'érythropoïétine endogène (EPO), la régulation des protéines
de transport du fer et la réduction de l'hepcidine
(une protéine régulatrice du fer augmentée pendant l'inflammation dans la MRC).
Cette réponse améliore la biodisponibilité du fer, augmente la production d'Hb et la masse érythrocytaire.
Effets
pharmacodynamiques
Effets sur
l'intervalle QTc et la fréquence cardiaque
Une étude
approfondie de l'intervalle QT conduite chez des sujets sains traités par roxadustat à une dose thérapeutique unique de 2,75 mg/kg et
une dose suprathérapeutique unique de 5 mg/kg (jusqu'à 510
mg) n'a pas montré d'allongement de l'intervalle QTc.
Cette même étude approfondie de l'intervalle QT a démontré une augmentation de
la fréquence cardiaque corrigée par placebo de 9 à 10 bpm entre 8 et 12 h après la dose de 2,75 mg/kg et de 15 à
18 bpm entre 6 et 12 h après la dose de 5 mg/kg.
Efficacité
et sécurité cliniques
Programme
de développement dans l'anémie associée à une MRC
L'efficacité
et la sécurité du roxadustat ont été évaluées pendant
au moins 52 semaines dans le cadre d'un programme de phase III mondial
constitué de 8 études multicentriques, randomisées conduites chez des patients
anémiés atteints d'une MRC non dépendants de la dialyse (NDD) et dépendants de
la dialyse (DD) (voir tableau 4).
Trois études menées chez des patients NDD atteints d'une MRC de stade 3-5 étaient en double aveugle versus placebo (ALPS, 1517-CL-0608 ; ANDES, FGCL-4592-060 ; OLYMPUS, D5740C00001) et une étude conduite en ouvert (DOLOMITES, 1517-CL-0610) utilisant la darbépoétine alfa comme comparateur. Toutes les études NDD ont évalué l'efficacité et la sécurité de patients non traités par ASE en corrigeant, puis en maintenant le taux d'Hb dans l'intervalle cible de 10 à 12 g/dL (études de correction du taux d'Hb).
Quatre études en ouvert, versus ASE (traitement contrôle : époétine alfa et/ou darbépoétine alfa), ont été conduites chez des patients hémodialysés ou sous dialyse péritonéale et ont évalué l'efficacité et la sécurité selon différentes approches :
Les patients
des études NDD étaient atteints d'une MRC de stade 3 à 5 et ne recevaient pas
de dialyse.
Tous les
patients avaient un taux moyen d'Hb ≤ 10,0 g/dL, excepté les patients de l'étude
DOLOMITES (1517-CL-0610), qui étaient autorisés à avoir un taux moyen d'Hb ≤ 10,5 g/dL.
Les taux de
ferritine devaient être ≥ 30 ng/mL (ALPS, 1517-CL-0608 ; ANDES, FGCL-4592-060), ≥ 50 ng/mL (OLYMPUS, D5740C00001) ou ≥
100 ng/mL (DOLOMITES,
1517-CL-0610). À l'exception
des patients de l'étude (OLYMPUS, D5740C00001), qui étaient autorisés à prendre
un traitement par ASE jusqu'à 6 semaines avant la randomisation, les patients
ne pouvaient pas recevoir de traitement par ASE dans les 12 semaines précédant
la randomisation.
Les patients des études DD devaient être dialysés : patients DD stables dans l'étude PYRENEES (1517-CL-0613), définis comme étant sous dialyse depuis plus de 4 mois ; ou patients DD incidents (ID) dans l'étude HIMALAYAS (FGCL-4592-063), définis comme étant sous dialyse depuis au moins 2 semaines et pas plus de 4 mois. Les patients des études SIERRAS (FGCL-4592-064) et ROCKIES (D5740C00002) étaient à la fois des patients DD stables (80 à 90 % environ) et ID (10 à 20 % environ). Tous les patients devaient avoir un taux de ferritine ≥ 100 ng/mL. Tous les patients étaient traités par un ASE par voie intraveineuse ou sous-cutanée depuis au moins 8 semaines avant la randomisation, à l'exception de l'étude HIMALAYAS (FGCL 4592063) ayant exclu les patients qui avaient reçu un traitement par ASE dans les 12 semaines précédant la randomisation.
Le traitement par roxadustat suivait les principes des instructions de prise décrites dans la rubrique Posologie et mode d'administration. Les données démographiques et toutes les caractéristiques d'inclusion de chaque étude étaient comparables entre les groupes roxadustat et les groupes contrôle. L'âge médian au moment de la randomisation était compris entre 55 et 69 ans, 16,6 à 31,1 % des patients se situant dans la tranche d'âge de 65 à 74 ans, et 6,8 à 35 % d'entre eux étant âgés de ≥ 75 ans. Le pourcentage de patientes était compris entre 40,5 et 60,7 %. Les races les plus fréquemment représentées sur l'ensemble des études étaient les races blanches, noires ou afro-américaines et asiatiques. Les étiologies de la MRC les plus courantes étaient la néphropathie hypertensive et diabétique. Les taux médians d'Hb variaient entre 8,60 et 10,78 g/dL. Environ 50 à 60 % des patients NDD et 80 à 90 % des patients DD avaient des réserves de fer suffisantes à l'inclusion.
Les données de sept études de phase III ont été réparties en deux populations distinctes (trois NDD et quatre DD) (voir tableau 4).
Trois études NDD versus placebo (2 386 patients sous roxadustat ; 1 884 patients sous placebo) ont été incluses dans le pool NDD. Les données de l'étude NDD DOLOMITES de phase III versus ASE (1517-CL-0610 ; 323 patients sous roxadustat et 293 patients sous darbépoétine alfa) ne sont pas incluses dans les analyses groupées des études NDD, car cette étude est menée en ouvert, versus comparateur actif.
Quatre études DD versus ASE (2 354 patients sous roxadustat ; 2 360 patients sous ASE [époétine alfa et/ou darbépoétine alfa]) ont été incluses dans le pool DD. Dans le pool DD, deux sous-pools ont été établis pour refléter les deux différentes approches thérapeutiques :
Tableau 4. Aperçu du programme de phase III de développement du roxadustat dans le traitement de l'anémie associée à une MRC
| Études menées chez des patients NDD | ||||
| Études versus placebo (pool NDD) | Etude versus ASE (darbépoétine alfa) | |||
| Approche | Correction du taux d'Hb | |||
| Étude | ALPS (1517-CL-0608) | ANDES (FGCL-4592-060) | OLYMPUS (D5740C00001) | DOLOMITES (1517-CL-0610) |
| Randomisée (roxadustat/ comparateur) | 594 (391/203) | 916 (611/305) | 2 760 (1384/1376) | 616 (323/293) |
| Études menées chez des patients DD | ||||
| Études versus ASE (pool DD) (époétine alfa ou darbépoétine alfa) | ||||
| Approche | Conversion ASE | Correction du taux d'Hb | Conversion ASE ou correction du taux d'Hb | |
| Étude | PYRENEES (1517-CL-0613) | SIERRAS (FGCL-4592-064) | HIMALAYAS (FGCL-4592-063) | ROCKIES (D5740C00002) |
| Randomisée (roxadustat/ comparateur) | 834 (414/420) | 740 (370/370) | 1 039 (522/517) | 2 101 (1 048/1 053) |
Patients atteints d'une MRC NDD Résultats d'efficacité
Évolution
du taux d'Hb pendant le traitement
Dans les
études cliniques, le roxadustat était efficace dans
l'obtention et le maintien des taux d'Hb cibles (10 à
12 g/dL) chez les patients anémiés atteints d'une MRC
non dialysés (voir figure 1).

Critères
d'efficacité principaux évalués par le taux d'Hb chez
des patients atteints d'une MRC NDD
Chez les patients NDD nécessitant un
traitement de leur anémie (correction du taux d'Hb),
la proportion de ceux ayant obtenu une réponse au cours des 24 premières
semaines était plus élevée dans le groupe roxadustat
(80,2 %) que dans le groupe placebo (8,7 %). Une augmentation statistiquement
significative du taux d'Hb a été observée entre
l'inclusion et les semaines 28 à 36 dans le groupe roxadustat
(1,91 g/dL) comparativement au groupe placebo (0,14
g/dL) et la limite inférieure de l'intervalle de
confiance à 95 % est supérieure à 1. Dans les études conduites chez des
patients NDD, une augmentation du taux d'Hb d'au
moins 1 g/dL a été obtenue, avec une durée médiane de
4,1 semaines (voir tableau 5).
Dans l'étude DOLOMITES (1517-CL-0610) en ouvert, versus ASE menée chez des patients NDD, la proportion de ceux ayant obtenu une réponse (correction du taux d'Hb) au cours des 24 premières semaines était non inférieure dans le groupe roxadustat (89,5 %) que dans le groupe darbépoétine alfa (78 %) (voir tableau 5).
Tableau 5. Critères d'efficacité principaux évalués par le taux d'Hb (NDD)
| Population | Patients atteints d'une MRC NDD | |||
| Approche | Correction du taux d'Hb | Correction du taux d'Hb | ||
| Critère/ Paramètre | Pool NDD (FAS) | DOLOMITES (PPS) 1517-CL-0610 | ||
| Roxadustat n = 2 368 | Placebo n = 1 865 | Roxadustat n = 286 | Darbépoétine alfa n = 273 | |
| Proportion de patients ayant obtenu une réponsea (correction du taux d'Hb) | ||||
| Répondeurs, n (%) [IC à 95 %] | 1 899 (80,2) [78,5 ; 81,8] | 163 (8,7) [7,5 ; 10,1] | 256 (89,5) [85,4 ; 92,8] | 213 (78,0) [72,6 ; 82,8] |
| Différence de proportions [IC à 95 %] | 71,5 [69,40 ; 73,51] | 11,51 [5,66 ; 17,36] | ||
| Odds ratio (rapport de cotes) [IC à 95 %] | 40,49 [33,01 ; 49,67] | 2,48 [1,53 ; 4,04] | ||
| Valeur de p | < 0,0001 | NE | ||
| Variation du taux d'Hb par rapport à l'inclusion (g/dL)b | ||||
| Taux moyen (DS) à l'inclusion | 9,10 (0,74) | 9,10 (0,73) | 9,55 (0,76) | 9,54 (0,69) |
| VPI moyenne (DS) | 1,85 (1,07) | 0,17 (1,08) | 1,85 (1,08) | 1,84 (0,97) |
| Population | Patients atteints d'une MRC NDD | |||
| Approche | Correction du taux d'Hb | Correction du taux d'Hb | ||
| Critère/ Paramètre | Pool NDD (FAS) | DOLOMITES (PPS) 1517-CL-0610 | ||
| Roxadustat n = 2 368 | Placebo n = 1 865 | Roxadustat n = 286 | Darbépoétine alfa n = 273 | |
| Moyenne des MC | 1,91 | 0,14 | 1,85 | 1,84 |
| Différence moyenne des MC [IC à 95 %] | 1,77 [1,69 ; 1,84] | 0,02 [-0,13 ; 0,16] | ||
| Valeur de p | < 0,0001 | 0,844 | ||
VPI : variation par rapport à l'inclusion ; IC : intervalle de confiance ; MRC : maladie rénale chronique ; FAS : ensemble d'analyse complet (full analysis set) ; Hb : hémoglobine ; MC : moindres carrés ; NE : non effectué ; NDD : non dépendant de la dialyse ; PPS : population per protocole : per protocol set) ; DS : déviation standard. aRéponse (correction du taux d'Hb) dans les 24 premières semaines. bVariation du taux d'Hb entre l'inclusion et les semaines 28 à 36.
Patients atteints d'une MRC DD
Évolution du taux d'Hb pendant le traitement
Dans les études cliniques, le roxadustat était efficace dans l'obtention et le maintien de taux d'Hb cibles (10 à 12 g/dL) chez les patients atteints d'une MRC dialysés, indépendamment de leur traitement antérieur par ASE (voir figures 2 et 3).

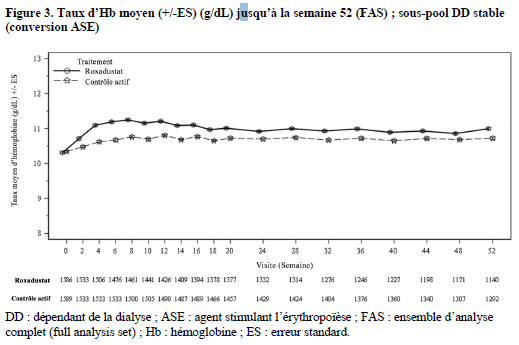
Critères
d'efficacité principaux évalués par le taux d'Hb chez
des patients atteints d'une MRC DD
Les patients DD nécessitant un traitement de
leur anémie pour corriger leur taux d'Hb et ceux
convertis à un traitement par ASE ont obtenu une augmentation de leur taux d'Hb entre l'inclusion et les semaines 28 à 36 dans le groupe
roxadustat ; cette augmentation était comparable à
celle observée dans le groupe ASE et supérieure à la marge de non-infériorité
prédéfinie de -0,75 g/dL. La proportion de patients
ayant obtenu une réponse sur leur taux d'Hb au cours
des 24 premières semaines était similaire dans les groupes roxadustat
et ASE (voir tableau 6).
Tableau 6. Critères d'efficacité principaux évalués par le taux d'Hb (DD)
| Population | Patients DD | |||
| Approche | Correction du taux d'Hb | Conversion ASE | ||
| Critère/ Paramètre | Pool DD ID (FAS/PPS) | Pool DD stable (PPS) | ||
| Roxadustat n = 756 | ASE n = 759 | Roxadustat n = 1 379 | ASE n = 1 417 | |
| Variation du taux d'Hb par rapport à l'inclusion (g/dL) | ||||
| Taux moyen (DS) à l'inclusion | 8,77 (1,20) | 8,82 (1,20) | 10,32 (0,99) | 10,37 (0,99) |
| VPI moyenne (DS) | 2,37 (1,57) | 2,12 (1,46) | 0,65 (1,15) | 0,36 (1,23) |
| Moyenne des MC | 2,17 | 1,89 | 0,58 | 0,28 |
| Différence moyenne des MC [IC à 95 %] | 0,28 [0,110 ; 0,451] | 0,30 [0,228 ; 0,373] | ||
| Valeur de p | 0,0013 | < 0,0001 | ||
| Proportion de patients ayant obtenu une réponse sur le taux d'Hb a,b | ||||
| Répondeurs, n (%) [IC à 95 %] | 453 (59,9) [56,3 ; 63,4] | 452 (59,6) [56,0 ; 63,1] | 978 (70,9) [68,4 ; 73,3] | 959 (67,7) [65,2 ; 70,1] |
| Différence de proportions [IC à 95 %] | 0,3 [-4,5 ; 5,1] | 2,7 [-0,7 ; 6,0] | ||
| Odds ratio (rapport de cotes) [IC à 95 %] | NE | NE | ||
| Valeur de p | NE | NE | ||
Traitement
de secours, transfusion de globules rouges (GR) et administration intraveineuse
de fer
Les effets du traitement par roxadustat sur
l'utilisation d'un traitement de secours, d'une transfusion de GR et sur
l'administration intraveineuse de fer sont présentés dans le tableau 7 (NDD) et
le tableau 8 (DD). Dans les études cliniques, le roxadustat
a diminué les taux sanguins d'hepcidine (régulateur
du mécanisme du fer), de la ferritine, a augmenté le fer sérique tandis que la
saturation de la transferrine est restée stable, tous ces paramètres ayant été
évalués au cours du temps en tant qu'indicateurs du statut martial.
Cholestérol
à lipoprotéines de faible densité (LDL cholestérol)
Les effets du
traitement par roxadustat sur le LDL cholestérol sont
présentés dans les tableaux 7 et 8. Les taux moyens de LDL cholestérol et de
HDL (lipoprotéines de haute densité) cholestérol ont diminué chez les patients
traités par roxadustat comparativement à ceux
recevant un placebo ou un ASE. L'effet sur le LDL cholestérol était plus
prononcé conduisant à une diminution du rapport LDL/HDL et ce, indépendamment
de l'utilisation de statines.
Tableau 7. Autres critères d'efficacité : recours à un traitement de secours, administration mensuelle intraveineuse de fer et variation par rapport à l'inclusion du LDL cholestérol (NDD)
| Population | Patients atteints d'une MRC NDD | |||
| Intervention | Correction | Correction | ||
| Critère/Paramètre | Pool NDD (FAS) | DOLOMITES (1517-CL-0610) | ||
| Roxadustat n = 2 368 | Placebo n = 1 865 | Roxadustat n = 322 | Darbépoétine alfa n = 292 | |
| Nombre de patients avec traitement de secours, n (%)* | 211 (8,9) | 580 (31,1) | NE | |
| Transfusion de GR | 118 (5,0) | 240 (12,9) | ||
| Fer IV | 50 (2,1) | 90 (4,8) | ||
| ASE | 48 (2,0) | 257 (13,8) | ||
| TI | 10,4 | 41,0 | ||
| Hazard ratio | 0,19 | NE | ||
| IC à 95 % | 0,16 ; 0,23 | |||
| Valeur de p | < 0,0001 | |||
| Nombre de patients traités par fer IV, n (%)† | NE | 20 (6,2) | 37 (12,7) | |
| TI | 9,9 | 21,2 | ||
| Hazard ratio | 0,45 | |||
| IC à 95 % | 0,26 ; 0,78 | |||
| Valeur de p | 0,004 | |||
| Variation du LDL cholestérol (mmol/L) entre l'inclusion et les semaines 12 à 28‡ | ||||
| Population | Patients atteints d'une MRC NDD | |||
| Intervention | Correction | Correction | ||
| Critère/Paramètre | Pool NDD (FAS) | DOLOMITES (1517-CL-0610) | ||
| Roxadustat n = 2 368 | Placebo n = 1 865 | Roxadustat n = 322 | Darbépoétine alfa n = 292 | |
| Analyse ANCOVA | ||||
| Moyenne des MC | -0,446 | 0,066 | -0,356 | 0,047 |
| IC à 95 % | -0,484 ; -0,409 | 0,017 ; 0,116 | -0,432 ; -0,280 | -0,033 ; 0,127 |
| Différence moyenne des MC (comparateur R) | -0,513 | -0,403 | ||
| IC à 95 % | -0,573 ; -0,453 | -0,510 ; -0,296 | ||
| Valeur de p | < 0,0001 | < 0,001 | ||
Tableau 8. Autres critères d'efficacité : recours à un traitement de secours, administration mensuelle intraveineuse de fer et variation par rapport à l'inclusion du LDL cholestérol (DD)
| Population | Patients atteints d'une MRC DD | |||
| Intervention | Correction | Conversion | ||
| Critère/ Paramètre | Pool DD ID (FAS) | Pool DD stable (FAS) | ||
| Roxadustat n = 756 | ASE n = 759 | Roxadustat n = 1 586 | ASE n = 1 589 | |
| Administration mensuelle moyenne de fer en IV sur les semaines 28 à 52 (mg)* | ||||
| n | 606 | 621 | 1 414 | 1 486 |
| Moyenne (ÉT) | 53,57 (143,097) | 70,22 (173,33) | 42,45 (229,80) | 61,99 (148,02) |
| Variation du LDL cholestérol (mmol/L) entre l'inclusion et les semaines 12 à 28 | ||||
| Analyse ANCOVA | ||||
| Moyenne des MC | -0,610 | -0,157 | -0,408 | -0,035 |
| IC à 95 % | -0,700 ; -0,520 | -0,245 ; -0,069 | -0,449 ; -0,368 | -0,074 ; 0,003 |
| Différence moyenne des MC (Comparateur R) | -0,453 | -0,373 | ||
| IC à 95 % | -0,575 ; -0,331 | -0,418 ; -0,328 | ||
| Valeur de p | < 0,0001 | < 0,0001 | ||
Dans l'étude SIERRAS (FGCL-4592-064) conduite chez des patients en dialyse, la proportion de patients ayant reçu une transfusion de globules rouges pendant le traitement était significativement plus faible dans le groupe roxadustat que dans le groupe EPO-alfa (12,5 % versus 21,1 %) ; la différence n'était pas statistiquement significative dans l'étude ROCKIES (D5740C00002) (9,8 % versus 13,2 %).
Résultats
rapportés par les patients non dialysés
Dans l'étude
DOLOMITES (1517-CL-0610), la non-infériorité du roxadustat
par rapport à la darbépoétine a été établie pour les
scores des échelles SF-36 PF et SF-36 VT.
Résultats
rapportés par les patients dialysés
Dans l'étude
PYRENEES (1517-CL-0613), la non-infériorité du roxadustat
par rapport à l'ASE a été établie concernant la variation des scores des
échelles SF-36 PF et SF-36 VT entre l'inclusion et les semaines 12 à 28.
Sécurité
clinique
Méta-analyse
des évènements cardiovasculaires adjudiqués et regroupés
Une
méta-analyse des évènements indésirables cardiovasculaires majeurs (MACE (Major
Adverse Cardiovascular Event) ; critère composite
incluant la mortalité toutes causes confondues [ACM], l'infarctus du myocarde
et l'AVC) et des MACE+ (critère composite incluant l'ACM, l'infarctus du
myocarde, l'AVC et l'hospitalisation pour un angor instable ou une insuffisance
cardiaque congestive) du programme d'études de phase III a été conduite chez 8
984 patients.
MACE, MACE+
et ACM dans l'ensemble de données des études contrôlées versus placebo
conduites sur la correction du taux d'Hb chez des
patients atteints d'une MRC NDD
Chez les
patients NDD, l'analyse des données MACE, MACE+ et ACM, provenant des analyses
sous traitement, incluait toutes les données depuis le début du traitement de l'étude
jusqu'à 28 jours après la fin du traitement (la fin du suivi). L'analyse sous
traitement a utilisé un modèle de Cox pondéré par l'inverse de la probabilité
d'être censuré (inverse probability of censoringweighting ou IPCW), qui
vise à corriger les différences de durée de suivi entre le roxadustat
et le placebo, y compris les facteurs identifiés contribuant à l'augmentation
du risque et à l'arrêt prématuré du traitement, en particulier les déterminants
du débit de filtration glomérulaire estimé (DFGe) et
le taux d'Hb à l'inclusion et au cours du temps. On
ne peut assurer qu'il ne demeure pas des facteurs de confusion dans ce modèle.
Les HR des analyses sous traitement étaient de 1,26, 1,17 et 1,16 (voir tableau
9). Les analyses ITT ont inclus toutes les données depuis le début du
traitement à l'étude jusqu'à la fin du suivi de la tolérance post- traitement.
Ces analyses ITT ont été incluses afin de refléter la distribution
déséquilibrée du risque en faveur du placebo dans l'analyse sous traitement. Toutefois,
une dilution de l'effet du traitement à l'étude est généralement observée dans
les analyses ITT. Un biais ne peut pas être complètement écarté, notamment en
raison de l'instauration du traitement de secours par un ASE après l'arrêt du
traitement à l'étude. Les HR étaient de 1,10, 1,07 et 1,08 avec des limites
supérieures des IC à 95 % de 1,27, 1,21 et 1,26 respectivement.
Tableau 9. Sécurité et mortalité CV dans le pool NDD de correction du taux d'Hbcontrolé versus placebo
| MACE | MACE+ | ACM | ||||
| Roxadustat n = 2 386 | Placebo n = 1 884 | Roxadustat n = 2 386 | Placebo n = 1 884 | Roxadustat n = 2 386 | Placebo n = 1 884 | |
| Sous traitement | ||||||
| Nombre de patients présentant des évènements (%) | 344 (14,4) | 166 (8,8) | 448 (18,8) | 242 (12,8) | 260 (10,9) | 122 (6,5) |
| FAIR | 8,7 | 6,8 | 11,6 | 10,1 | 6,4 | 5,0 |
| HR (IC à 95 %) | 1,26 (1,02 ; 1,55) | 1,17 (0,99 ; 1,40) | 1,16 (0,90 ; 1,50) | |||
| ITT | ||||||
| Nombre de patients avec évènement (%) | 480 (20,1) | 350 (18,6) | 578 (24,2) | 432 (22,9) | 400 (16,8) | 301 (16) |
| FAIR | 10,6 | 10,3 | 13,2 | 13,2 | 8,3 | 8,1 |
| HR (IC à 95 %) | 1,10 (0,96 ; 1,27) | 1,07 (0,94 ; 1,21) | 1,08 (0,93 ; 1,26) | |||
MACE, MACE+
et ACM dans l'ensemble de données des études contrôlées versus ASE conduites
sur la correction du taux d'Hb chez des patients
atteints d'une MRC NDD et chez des patients ID-DD
Dans l'intervention de
correction du taux d'Hb des patients NDD et ID-DD,
les caractéristiques à l'inclusion et les taux d'arrêt du traitement étaient
comparables entre les patients sous roxadustatpoolés et ceux sous ASE poolés.
L'analyse des données MACE, MACE+ et ACM, observées sous traitement ont montré
des HR de 0,79, 0,78 et 0,78, avec des limites supérieures des IC à 95 % de
1,02, 0,98 et 1,05, respectivement (voir tableau 10). Les analyses des données
sous traitement confirment l'absence de preuves d'une augmentation du risque en
termes de tolérance cardiovasculaire ou de mortalité avec le roxadustat par rapport à un ASE chez les patients atteints
d'une MRC nécessitant une correction de leur taux d'Hb.
Tableau 10. Sécurité et mortalité CV dans le pool de correction du taux d'Hb contrôlé versus ASE
| MACE | MACE+ | ACM | ||||
| Roxadustat n = 1 083 | ASE n = 1 059 | Roxadustat n = 1 083 | ASE n = 1 059 | Roxadustat n = 1 083 | ASE n = 1 059 | |
| Sous traitement | ||||||
| Nombre de patients présentant des évènements (%) | 105 (9,7) | 136 (12,8) | 134 (12,4) | 171 (16,1) | 74 (6,8) | 99 (9,3) |
| TI | 6,5 | 8,2 | 8,3 | 10,3 | 4,6 | 6,0 |
| HR (IC à 95 %) | 0,79 (0,61 ; 1,02) | 0,78 (0,62 ; 0,98) | 0,78 (0,57 ; 1,05) | |||
MACE, MACE+
et ACM dans l'ensemble de données des études contrôlées versus ASE conduites
sur la conversion ASE chez des patients atteints d'une MRC stable DD
Chez les
patients DD stables convertis d'un ASE au roxadustat,
les résultats de l'analyse des critères MACE, MACE+ et ACM observés sous
traitement ont montré des HR de 1,18, 1,03 et 1,23, avec des limites
supérieures des IC à 95 % pour les HR de 1,38, 1,19 et 1,49, respectivement
(voir tableau 11). Les résultats du tableau 11 doivent être interprétés avec
prudence, car pour les patients qui ont été convertis d'un traitement par ASE
au traitement par roxadustat au début de l'étude,
l'impact d'un risque inhérent au passage à un nouveau traitement par rapport au
maintien du traitement existant avec un taux d'Hb
stabilisé peut fausser les résultats observés et par conséquent toute
comparaison de l'effet du traitement estimé ne peut être établie avec
fiabilité.
Tableau 11. Sécurité et mortalité CV dans le pool DD stable de conversion ASE versus ASE
| MACE | MACE+ | ACM | ||||
| Roxadustat n = 1 594 | ASE n = 1 594 | Roxadustat n = 1 594 | ASE n = 1 594 | Roxadustat n = 1 594 | ASE n = 1 594 | |
| Sous traitement | ||||||
| Nombre de patients avec évènements (%) | 297 (18,6) | 301 (18,9) | 357 (22,4) | 403 (25,3) | 212 (13,3) | 207 (13,0) |
| TI | 10,4 | 9,2 | 12,5 | 12,3 | 7,4 | 6,3 |
| HR (IC à 95 %) | 1,18 (1,00 ; 1,38) | 1,03 (0,90 ; 1,19) | 1,23 (1,02 ; 1,49) | |||
L'exposition plasmatique au roxadustat (aire sous la courbe de la concentration plasmatique du médicament dans le temps [ASC] et les concentrations plasmatiques maximales [Cmax]) est proportionnelle à la dose dans un intervalle de doses thérapeutiques recommandées. Dans le schéma de prise trois fois par semaine, les concentrations plasmatiques de roxadustat à l'état d'équilibre sontobtenues en une semaine (3 doses) avec une accumulation minimale. La pharmacocinétique du roxadustat ne varie pas dans le temps.
Absorption
Les concentrations plasmatiques maximales (Cmax) sont généralement atteintes 2 heures après la prise de la dose à jeun.
La prise de roxadustat avec de la nourriture a diminué la Cmax
de 25 % mais n'a pas modifié l'ASC par rapport à une prise à jeun. Par
conséquent, le roxadustat peut être pris avec ou sans nourriture (voir
rubrique Posologie et mode d'administration).
Distribution
Le roxadustat a une forte affinité de liaison avec les protéines
plasmatiques humaines (environ 99 %), principalement l'albumine. Le
rapport sang/plasma du roxadustat est de 0,6. Le volume apparent de
distribution à l'état d'équilibre est de 24 L.
Biotransformation
Sur la base des données in vitro, le roxadustat est un substrat
des enzymes CYP2C8 et UGT1A9, ainsi que de la BCRP, l'OATP1B1, l'OAT1
et l'OAT3. Le roxadustat n'est pas un substrat de l'OATP1B3 ni de la
P-gp. Le roxadustat est principalement métabolisé en hydroxy-roxadustat
et roxadustat-O-glucuronide. Le roxadustat sous forme inchangée
était le principal composant circulant dans le plasma humain ; aucun
métabolite décelable dans le plasma humain n'a constitué plus de 10 %
de l'exposition totale à des substances liées au médicament et aucun
métabolite spécifique à l'homme n'a été observé.
Élimination
La demi-vie efficace moyenne (t1/2) du roxadustat est d'environ 15 heures chez les patients atteints d'une MRC.
La clairance corporelle apparente totale (CL/F) du roxadustat est de
1,1 L/h chez les patients atteints d'une MRC non dialysés et de 1,4 L/h
chez les patients atteints d'une MRC dialysés. Le roxadustat et ses
métabolites ne sont pas significativement éliminés par hémodialyse.
Lors de l'administration par voie orale de roxadustat radiomarqué à des sujets sains, en moyenne, 96 % de la radioactivité a été éliminée (50 % dans les fèces, 46 % dans l'urine). Dans les fèces, 28 % de la dose était excrétée sous forme de roxadustat inchangé. Moins de 2 % de la dose de roxadustat était retrouvée sous forme inchangée dans l'urine.
Populations particulières
Effets de l'âge, du sexe, du poids corporel et de la race
Aucune différence cliniquement pertinente de la pharmacocinétique du
roxadustat n'a été observée en fonction de l'âge (≥ 18 ans), du sexe,
de la race, du poids corporel, de la fonction rénale (DFGe) ou du
statut en dialyse chez des patients adultes présentant une anémie
associée à une MRC.
Hémodialyse
Chez les patients atteints d'une MRC dépendants de la dialyse, aucune
différence marquée des valeurs des paramètres pharmacocinétiques n'a
été observée lorsque le roxadustat était administré 2 heures avant ou 1
heure après l'hémodialyse. La dialyse est une voie négligeable de
clairance globale du roxadustat.
Insuffisance hépatique
Après une dose unique de 100 mg de roxadustat, l'ASC moyenne du roxadustat était augmentée de 23 % et la Cmax
moyenne diminuée de 16 % chez les sujets présentant une insuffisance
hépatique modérée (Child-Pugh classe B) et une fonction rénale normale,
comparativement aux sujets dont les fonctions hépatiques et rénales
étaient normales. Chez les sujets présentant une insuffisance hépatique
modérée (Child-Pugh classe B) et une fonction rénale normale, l'ASCinf du roxadustat non lié était augmentée (+70 %) par rapport aux sujets sains.
La pharmacocinétique du roxadustat chez les sujets présentant une
insuffisance hépatique sévère (Child-Pugh classe C) n'a pas été étudiée.
Interactions médicamenteuses
Sur la base des données in vitro, le roxadustat est un inhibiteur du CYP2C8, de la BCRP, de l'OATP1B1 et de l'OAT3 (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
La pharmacocinétique de la rosiglitazone (substrat sensible modéré du
CYP2C8) n'a pas été affectée par la coadministration de roxadustat. Le
roxadustat peut être un inhibiteur de l'UGT1A1 intestinal mais pas de
l'UGT1A1 hépatique et n'a pas fait la preuve d'une inhibition d'autres
transporteurs ou enzymes métabolisant le CYP, ni d'une induction
d'enzymes du CYP, aux concentrations cliniquement pertinentes. Le
charbon activé oral ou l'oméprazole n'ont aucun effet cliniquement
significatif sur la pharmacocinétique du roxadustat. Le clopidogrel n'a
pas d'effet sur l'exposition au roxadustat chez les patients atteints
d'une MRC.
Le roxadustat a une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines. Des cas de convulsions ont été rapportés pendant le traitement par Evrenzo (voirrubrique Mises en garde spéciales et précautions d'emploi). Par conséquent, la prudence est de mise lors de la conduite de véhicules ou l'utilisation de machines.
Études de toxicologie en administration répétée
Dans l'étude en administration répétée intermittente de 26 semaines
menée chez des rats Sprague-Dawley ou Fisher, le roxadustat, administré
à environ 4 à 6 fois l'ASC totale à la dose humaine maximale
recommandée (DHMR), a conduit à des résultats histopathologiques
anormaux, dont des valvulopathies des valves aortiques et
auriculoventriculaires (A-V). Ces résultats étaient présents chez les
animaux survivants au moment de l'arrêt de l'étude, de même que chez
les animaux sortis prématurément de l'étude dans un état moribond. En
outre, ils n'ont pas été totalement réversibles car ils étaient encore
présents chez les animaux à la fin de la période de récupération de 30
jours.
Des études de toxicité en administration répétée menées chez des animaux sains ont montré qu'une pharmacologie exagérée conduisait à une érythropoïèse excessive.
Des modifications hématologiques telles que des diminutions des plaquettes circulantes et des allongements du temps de céphaline activée et du temps de prothrombine ont été enregistrées chez des rats à environ 2 fois l'ASC totale à la DHMR. Des thrombus ont été observés dans la moelle osseuse (expositions systémiques d'environ 7 fois l'ASC totale à la DHMR chez des rats), dans les reins (expositions systémiques d'environ 5 à 6 fois l'ASC totale à la DHMR chez des rats), dans les poumons (expositions systémiques d'environ 8 et 2 fois l'ASC totale à la DHMR chez des rats et des singes cynomolgus, respectivement) et dans le cœur (expositions systémiques d'environ 4 à 5 fois l'ASC totale à la DHMR chez des rats).
Tolérance cérébrale
Dans l'étude en administration répétée intermittente de 26 semaines
menée chez des rats Sprague-Dawley, un animal a présenté un résultat
histologique anormal avec une nécrose et une gliose cérébrale, à
environ 6 fois l'ASC totale à la DHMR. Chez les rats Fisher, pour
lesquels la durée du traitement était la même, une nécrose cérébrale/de
l'hippocampe a été notée chez un total de quatre animaux, à environ 3 à
5 fois l'ASC totale à la DHMR.
Des singes cynomolgus, qui ont reçu des doses intermittentes de roxadustat pendant 22 ou 52 semaines, n'ont pas présenté de résultats similaires à des expositions systémiques allant jusqu'à 2 fois l'ASC totale à la DHMR.
Carcinogénicité et mutagénicité
Le roxadustat s'est révélé négatif dans le test in vitro de mutagénicité d'Ames, le test in vitro d'aberration chromosomique dans des lymphocytes du sang périphérique humain et le test in vivo du micronoyau chez la souris, à 40 fois la DHMR basée sur une dose équivalente humaine.
Dans les études de carcinogénicité conduites chez le rat et la souris, les animaux ont reçu du roxadustat selon un schéma d'administration de trois fois par semaine. Du fait de la clairance rapide du roxadustat chez les rongeurs, les expositions systémiques n'étaient pas continues pendant toute la période d'administration. De possibles effets carcinogéniques non ciblés peuvent par conséquent être sous-estimés.
Dans l'étude de carcinogénicité de 2 ans conduite chez des souris, des augmentations significatives de l'incidence du carcinome bronchoalvéolaire ont été observées dans les groupes à dose faible et élevée (expositions systémiques d'environ 1 fois et d'environ 3 fois l'ASC totale à la DHMR). Une augmentation significative du fibrosarcome sous-cutané a été observée chez les femelles du groupe à dose élevée (expositions systémiques d'environ 3 fois l'ASC totale à la DHMR).
Dans l'étude de carcinogénicité de 2 ans conduite chez des rats, une augmentation significative de l'incidence de l'adénome mammaire a été observée à la dose moyenne (exposition systémique d'environ 1 fois l'ASC totale à la DHMR). Toutefois, ce résultat n'était pas dose dépendant et l'incidence de ce type de tumeur était plus faible à la dose la plus élevée testée (exposition systémique d'environ 2 fois l'ASC totale à la DHMR) et n'a par conséquent pas été considérée comme liée au produit étudié.
Les études cliniques n'ont pas montré de résultats similaires à ceux des études de carcinogénicité conduites chez la souris et le rat.
Toxicité sur la reproduction et le développement
Le roxadustat n'a eu aucun effet sur la reproduction ou la fertilité
des rats mâles ou femelles traités, à environ 4 fois l'exposition
humaine à la DHMR. Cependant, à la DSEIO (dose sans effet indésirable
observé) chez des rats mâles, des diminutions du poids de l'épididyme
et des vésicules séminales (avec liquide) ont été observées sans effet
sur la fertilité mâle. La DSEO (dose sans effet observable) pour tous
les résultats liés aux organes de la reproduction chez le mâle
équivalait à 1,6 fois la DHMR. Chez les rats femelles, le nombre
d'embryons non viables et de pertes post-implantation était augmenté à
ce niveau de dose par rapport aux animaux contrôles.
Les résultats des études de toxicité sur la reproduction et le développement conduites chez les rats et les lapins ont démontré une réduction du poids moyen corporel fœtal et de la progéniture, une augmentation du poids placentaire moyen, des avortements et de la mortalité de la progéniture.
Des rates Sprague-Dawley gravides auxquelles du roxadustat a été administré quotidiennement du moment de l'implantation jusqu'à la soudure du palais dur (gestation des jours 7 à 17) ont présenté une diminution du poids corporel fœtal et une augmentation des altérations squelettiques à environ6 fois l'ASC totale à la DHMR. Le roxadustat n'a aucun effet sur la survie fœtale après implantation.
Des lapines New Zealand gravides ont reçu du roxadustat quotidiennement des jours 7 à 19 de gestation et des césariennes ont été pratiquées au jour 29 de la gestation. L'administration de roxadustat à des expositions systémiques allant jusqu'à environ 3 fois l'ASC totale à la DHMR n'a pas montré d'altération embryo-fœtale. Toutefois, une lapine a avorté à environ 1 fois l'ASC totale à la DHMR et 2 lapines ont avorté à environ 3 fois l'ASC totale à la DHMR. Les femelles ayant avorté étaient maigres.
Dans l'étude du développement périnatal/postnatal conduite chez des rats Sprague-Dawley, les rates gravides ont reçu du roxadustat quotidiennement du jour 7 de la gestation au jour 20 de la lactation. Durant la période de lactation, la mortalité des ratons des rates ayant reçu le roxadustat à environ2 fois la C max totale à la DHMR était élevée pendant la période pré-sevrage et les ratons ont été sacrifiés au moment du sevrage. Les ratons ayant reçu le roxadustat à des doses conduisant à des expositions systémiques d'environ 3 fois l'exposition humaine à la DHMR ont présenté une diminution significative de la survie 21 jours après la naissance (indice de lactation) comparativement aux ratons des portées contrôles.
Dans une étude d'adoption croisée, les effets les plus prononcés sur la viabilité des ratons ont été observés chez ceux exposés au roxadustat après la naissance uniquement, et la viabilité des ratons exposés au roxadustat jusqu'à la naissance était plus faible que chez ceux non exposés.
L'étude d'adoption croisée, dans laquelle des ratons de rates non exposées ont été nourris par des rates traitées par le roxadustat (dose humaine équivalente d'environ 2 fois la DHMR), a montré la présence de roxadustat dans le plasma des ratons, témoignant du passage du médicament dans le lait. Le lait de ces rates contenait du roxadustat. Les ratons qui étaient exposés au lait contenant du roxadustat ont présenté un taux de survie plus faible (85,1 %) que ceux nés de mères non traitées et nourris par des rates non traitées (taux de survie de 98,5 %). Le poids corporel moyen des ratons survivants exposés au roxadustat pendant la période de lactation était également plus faible que celui des ratons contrôles (qui n'avaient été exposés ni in utero ni par le lait).
Tolérance cardiovasculaire
Une étude de pharmacologie de sécurité cardiovasculaire a montré que la
fréquence cardiaque augmente après une seule administration de 100
mg/kg de roxadustat à des singes. Aucun effet n'a été observé sur
l'hERG ou l'ECG. Des études pharmacologiques de sécurité
supplémentaires conduites chez des rats ont montré que le roxadustat
réduisait la résistance périphérique totale, suivie d'une augmentation
réflexe de la fréquence cardiaque à environ 6 fois l'exposition à la
DHMR.
Pas d'exigences particulières pour l'élimination.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Prescription initiale hospitalière annuelle.
Prescription réservée aux spécialistes et services
NEPHROLOGIE.
Remboursement en fonction de l'indication (JO du 13/12/2022) :
La seule indication thérapeutique ouvrant droit à la prise en charge ou au remboursement par l'assurance maladie est :
- traitement de l'anémie symptomatique associée à une maladie rénale
chronique uniquement chez les patients adultes qui ne sont pas déjà
traités par un agent stimulant l'érythropoïèse (ASE), non dialysés ou
dialysés depuis moins de 4 mois.
Comprimés pelliculés (comprimés).
Comprimés rouges, en forme d'amande (d'environ 14 mm × 9 mm) portant l'inscription « 150 » sur une face.
Plaquettes thermoformées perforées pour délivrance à l'unité en PVC/aluminium dans un emballage contenant 12 x 1 comprimés pelliculés.
Chaque comprimé contient 150 mg de roxadustat.
Excipient(s) à effet notoire :
Chaque comprimé pelliculé de 150 mg contient 303,5 mg de lactose, 3,7
mg de laque d'aluminium rouge Allura AC et 0,84 mg de lécithine de soja.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Noyau du comprimé
Lactose monohydraté
Cellulose microcristalline (E460 (i))
Croscarmellose sodique (E468)
Povidone (E1201)
Stéarate de magnésium (E470b)
Pelliculage
Alcool polyvinylique (E1203)
Talc (E553b)
Macrogol (E1521)
Laque d'aluminium rouge Allura AC (E129)
Dioxyde de titane (E171)
Lécithine (soja) (E322)