
FORXIGA 10 mg, comprimé pelliculé, boîte de 30 plaquettes thermoformées de 1
Retiré du marché le : 27/10/2021
Dernière révision : 07/07/2021
Taux de TVA : 0%
Laboratoire exploitant : ASTRAZENECA
Source :

Forxiga est indiqué dans le traitement de la maladie rénale chronique chez l'adulte :
• En association à un traitement standard optimisé (inhibiteur de l'enzyme de conversion (IEC) ou antagoniste du récepteur de l'Angiotensine II (sartans), sauf si contre-indication)
• Avec un débit de filtration glomérulaire (DFG) compris entre 25 et 75 mL/min/1,73m2 et un rapport albumine / créatinine urinaire compris entre 200 et 5 000 mg/g
• Insuffisamment contrôlée malgré des thérapeutiques médicamenteuses bien conduites : IEC ou sartans.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
L'expérience de l'initiation d'un traitement par la dapagliflozine chez les patients avec un DFG < 25 mL/min est limitée.
L'efficacité glycémique de la dapagliflozine dépend de la fonction rénale et l'efficacité est réduite chez les patients avec un DFG < 45 mL/min et est vraisemblablement absente chez les patients atteints d'insuffisance rénale sévère (voir rubriques Posologie et mode d'administration, Propriétés pharmacodynamiques et Propriétés pharmacocinétiques).
Dans une étude chez des patients avec un diabète de type 2 atteints d'insuffisance rénale modérée (DFG < 60 mL/min), une plus forte proportion de patients traités par dapagliflozine a présenté des effets indésirables à type d'élévation de l'hormone parathyroïdienne (PTH) et d'hypotension comparé au placebo.
Insuffisance hépatique
L'expérience des études cliniques chez les patients atteints d'une insuffisance hépatique est limitée. L'exposition à la dapagliflozine est augmentée chez les patients atteints d'une insuffisance hépatique sévère (voir rubriques Posologie et mode d'administration et Propriétés pharmacocinétiques).
Utilisation chez les patients à risque de déplétion volémique et/ou d'hypotension
En raison de son mécanisme d'action, la dapagliflozine augmente la diurèse, ce qui pourrait être à l'origine de la baisse modérée de la pression artérielle observée dans les études cliniques (voir rubrique Propriétés pharmacodynamiques). Celle- ci pourrait être plus prononcée chez les patients avec des glycémies très élevées.
Une attention particulière devra être portée aux patients pour lesquels une baisse de la pression artérielle induite par la dapagliflozine peut représenter un risque, comme les patients sous traitement antihypertenseur avec un antécédent d'hypotension ou les patients âgés.
En cas de conditions intercurrentes qui peuvent entraîner une déplétion volémique (par exemple : une maladie gastro-intestinale), une surveillance attentive de l'état d'hydratation (par exemple : examen clinique, mesure de la pression artérielle, bilans biologiques incluant l'hématocrite et les électrolytes) est recommandée. Une interruption temporaire du traitement par dapagliflozine est recommandée chez les patients qui développent une déplétion volémique jusqu'à correction de la déplétion (voir rubrique Effets indésirables).
Acidocétose diabétique
Les inhibiteurs du co-transporteur de sodium-glucose de type 2 (SGLT2) doivent être utilisés avec prudence chez les patients présentant un risque accru d'ACD. Les patients qui peuvent être à risque accru d'ACD incluent les patients avec une faible réserve de cellules bêta fonctionnelles (p. ex. les patients avec un diabète de type 1, les patients avec un diabète de type 2 avec peu de peptides C ou un diabète auto-immun latent de l'adulte (LADA) ou les patients avec un antécédent de pancréatite), les patients dont les états conduisent à une absorption alimentaire réduite ou à une déshydratation sévère, les patients chez qui les doses d'insuline sont réduites et les patients avec des besoins accrus en insuline en raison d'une affection médicale aiguë, d'une intervention chirurgicale ou d'une consommation excessive d'alcool.
Le risque d'acidocétose diabétique doit être envisagé en cas de survenue de symptômes non spécifiques tels que : nausées, vomissements, anorexie, douleurs abdominales, soif intense, difficulté à respirer, confusion, fatigue inhabituelle ou somnolence. Si ces symptômes apparaissent, il faut immédiatement rechercher une acidocétose chez ces patients, indépendamment de la glycémie.
Avant d'initier la dapagliflozine, il faut tenir compte des facteurs pouvant prédisposer à une acidocétose dans les antécédents médicaux du patient.
Le traitement doit être interrompu chez les patients qui sont hospitalisés pour des interventions chirurgicales lourdes ou des pathologies médicales aiguës graves. La surveillance des corps cétoniques est recommandée chez ces patients. Le contrôle de la cétonémie (taux de cétone dans le sang) est préféré à la cétonurie (taux de cétone dans l'urine). Le traitement par la dapagliflozine ne peut être repris que quand les taux de corps cétoniques sont normaux et après une stabilisation de l'état du patient.
Diabète de type 2
De rares cas d'ACD, incluant des cas ayant conduit à la mise en jeu du pronostic vital et des cas d'issue fatale, ont été rapportés chez des patients traités par des inhibiteurs du SGLT2, y compris avec la dapagliflozine. Dans un certain nombre de cas, le tableau clinique était de présentation atypique, avec seulement une élévation modérée de la glycémie, en dessous de 14 mmol/L (250 mg/dL).
Pour les patients chez qui une ACD est suspectée ou diagnostiquée, le traitement par dapagliflozine doit être immédiatement arrêté.
La reprise d'un traitement par inhibiteurs du SGLT2 chez les patients présentant une ACD sous traitement par inhibiteurs du SGLT2 n'est pas recommandée sauf si un autre facteur déclenchant est identifié et corrigé.
Diabète de type 1
La dapagliflozine n'a pas été étudiée dans le traitement de l'insuffisance cardiaque ou de la maladie rénale chronique chez les patients présentant un diabète de type 1. Le traitement avec la dapagliflozine 10 mg n'est pas recommandé chez ces patients.
Fasciite nécrosante du périnée (gangrène de Fournier)
Des cas de fasciite nécrosante du périnée (aussi appelée « gangrène de Fournier ») survenus après mise sur le marché ont été rapportés chez des patients de sexe masculin et féminin prenant des inhibiteurs du SGLT2 (voir rubrique Effets indésirables). Cet événement rare mais grave et mettant potentiellement en jeu le pronostic vital des patients nécessite une intervention chirurgicale et un traitement antibiotique en urgence.
Il convient de recommander aux patients de consulter un médecin s'ils développent des symptômes tels qu'une douleur, une sensibilité, un érythème ou une tuméfaction au niveau de la zone génitale ou périnéale, accompagnés de fièvre ou de malaises. Il convient de garder à l'esprit que la fasciite nécrosante peut être précédée d'une infection urogénitale ou d'un abcès périnéal. N cas de suspicion de gangrène de Fournier, le traitement par Forxiga doit être interrompu et un traitement rapide (comprenant des antibiotiques et un débridement chirurgical) doit être instauré.
Infections des voies urinaires
L'excrétion urinaire de glucose peut être associée à un risque accru d'infection des voies urinaires ; une interruption temporaire de la dapagliflozine doit donc être envisagée lors du traitement d'une pyélonéphrite ou d'un sepsis urinaire.
Les patients âgés peuvent avoir un risque plus important de déplétion volémique et sont plus susceptibles d'être traités par des diurétiques.
Les patients âgés sont plus susceptibles d'avoir une altération de la fonction rénale et/ou d'être traités par des médicaments antihypertenseurs qui peuvent provoquer des modifications de la fonction rénale tels que les inhibiteurs de l'enzyme de conversion de l'angiotensine (IEC) et les antagonistes des récepteurs de l'angiotensine II de type 1 (ARA II). Les mêmes recommandations que celles pour la fonction rénale s'appliquent aux patients âgés comme à tous les autres patients (voir rubriques Posologie et mode d'administration, Mises en garde et précautions d'emploi, Effets indésirables et Propriétés pharmacodynamiques).
Insuffisance cardiaque
L'expérience de la dapagliflozine chez les patients de classe fonctionnelle NYHA IV est limitée.
Bilan urinaire
En raison de son mécanisme d'action, les patients prenant Forxiga auront un test de glucose urinaire positif.
Lactose
Les comprimés contiennent du lactose. Les patients atteints de troubles héréditaires rares d'intolérance au galactose, de déficit total en lactase ou de malabsorption du glucose-galactose ne doivent pas prendre ce médicament.
Résumé du profil de sécurité
Diabète de type 2
Dans les études cliniques conduites dans le diabète de type 2, plus de 15 000 patients ont été traités par dapagliflozine.
L'évaluation principale de sécurité d'emploi et de tolérance a été réalisée dans le cadre d'une analyse poolée préspécifiée de 13 études à court terme (jusqu'à 24 semaines) contrôlées versus placebo avec 2 360 patients traités par dapagliflozine 10 mg et 2 295 par placebo.
Dans l'étude des effets cardiovasculaires conduite avec la dapagliflozine, dans le diabète de type 2 (étude DECLARE, voir rubrique Propriétés pharmacodynamiques), 8 574 patients ont reçu de la dapagliflozine 10 mg et 8 569 ont reçu un placebo pendant une durée d'exposition médiane de 48 mois. En tout, il y a eu 30 623 patients-années d'exposition à la dapagliflozine.
Les effets indésirables les plus fréquemment rapportés dans les études cliniques étaient les infections génitales.
Diabète de type 1
Dans le cadre de deux études contrôlées versus placebo conduites chez des patients atteints de diabète de type 1, 548 patients ont été traités par dapagliflozine 5 mg en association avec une insulinothérapie ajustable et 532 ont reçu un placebo en association avec une insulinothérapie ajustable.
Le profil de sécurité de la dapagliflozine chez les patients présentant un diabète de type 1 était similaire au profil de sécurité de la dapagliflozine chez les patients présentant un diabète de type 2. Chez les patients présentant un diabète de type 1, les cas d'acidocétose diabétique étaient reportés comme fréquents. Voir « Description de certains effets indésirables » et rubrique Mises en garde et précautions d'emploi.
Insuffisance cardiaque
Dans l'étude des effets cardiovasculaires conduite avec la dapagliflozine chez des patients atteints d'insuffisance cardiaque à fraction d'éjection réduite (étude DAPA-HF), 2 368 patients ont été traités par la dapagliflozine à la dose de 10 mg et 2 368 patients ont reçu un placebo pendant une durée d'exposition médiane de 18 mois. La population de patients incluait des patients diabétiques de type 2 ou non diabétiques et des patients avec un DFGe ≥ 30 mL/min/1,73 m2.
Le profil de sécurité global de la dapagliflozine chez les patients atteints d'insuffisance cardiaque était cohérent avec le profil de sécurité connu de la dapagliflozine.
Maladie rénale chronique
Dans l'étude des effets rénaux conduite avec la dapagliflozine chez des patients atteints de maladie rénale chronique (DAPA-CKD), 2 149 patients ont été traités par la dapagliflozine à la dose de 10 mg et 2 149 patients ont reçu un placebo pendant une durée d'exposition médiane de 27 mois. La population de patients incluait des patients diabétiques de type 2 ou non diabétiques, avec un DFGe ≥ 25 à ≤ 75 mL/min/1,73 m². Le traitement était poursuivi si le DFGe diminuait à moins de 25 mL/min/1,73 m².
Le profil de sécurité global de la dapagliflozine chez les patients atteints de maladie rénale chronique était cohérent avec le profil de sécurité connu de la dapagliflozine.
Liste tabulée des effets indésirables
Les effets indésirables suivants ont été identifiés dans les études cliniques contrôlées versus placebo et lors de la surveillance en post-commercialisation. Aucun ne s'est révélé dose dépendant. Les effets indésirables mentionnés ci-dessous sont classés par fréquence et par classe de systèmes d'organes (SOC). Les différentes catégories de fréquence adoptent la convention suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
| Classe de systèmes d'organes | Très fréquent | Fréquent* | Peu fréquent** | Rare | Très rare |
| Infections et infestations | | Vulvovaginite, balanite et infections génitales associées*,b,c Infection des voies urinaires*,b,d | Infection fongique** | | Fasciite nécrosante du périnée (gangrène de Fournier)b,i |
| Troubles du métabolisme et de la nutrition | Hypoglycémie (quand utilisé avec SU ou insuline)b | | Déplétion volémiqueb,e Soif** | Acidocétose diabétique (en cas d'utilisation dans le diabète de type 2)b,i,k | |
| Affections du système nerveux | | Sensations vertigineuses | | | |
| Affections gastro-intestinales | | | Constipation** Sécheresse buccale** | | |
| Affections de la peau et du tissu sous-cutané | | Rashj | | | Angio- oedème |
| Affections musculo-squelettiq ues et systémiques | | Douleur dorsale* | | | |
| Affections du rein et des voies urinaires | | Dysurie Polyurie*,f | Nycturie** | | |
| Affections des organes de reproduction et du sein | | | Prurit vulvo vaginal** Prurit génital** | | |
| Investigations | | Augmentation de l'hématocriteg Diminution de la clairance rénale de la créatinine pendant le traitement initialb | Elévation de la créatininémie pendant le traitement initial**,b Elévation de l'urémie** Perte de poids** | | |
| Dyslipidémieh |
a Le tableau présente des données recueillies sur 24 semaines (court terme), n'excluant pas l'administration d'un traitement antidiabétique de secours.
b Voir paragraphe correspondant ci-dessous pour plus d'informations.
c La vulvovaginite, la balanite et les infections génitales associées incluent, par exemple les termes recommandés prédéfinis : infection mycosique vulvo vaginale, infection vaginale, balanite, infection génitale fongique, candidose vulvo vaginale, vulvovaginite, balanite candidosique, candidose génitale, infection génitale, infection génitale masculine, infection pénienne, vulvite, vaginite bactérienne, abcès vulvaire.
d L'infection des voies urinaires inclut les termes préférés suivants, mentionnés par ordre de fréquence rapportée : infection des voies urinaires, cystite, infection des voies urinaires par Escherichia, infection des voies génito-urinaires, pyélonéphrite, trigonite, uréthrite, infection rénale et prostatite.
e La déplétion volémique regroupe, par exemple, les termes recommandés prédéfinis suivants : déshydratation, hypovolémie, hypotension.
f La polyurie regroupe les termes préférés suivants : pollakiurie, polyurie, augmentation du volume urinaire
g Les variations moyennes par rapport à la valeur initiale de l'hématocrite étaient 2,30 % pour dapagliflozine 10 mg versus 0,33 % pour le placebo. Des valeurs de l'hématocrite >55 % ont été rapportées chez 1,3 % des sujets traités par dapagliflozine 10 mg versus 0,4 % des sujets recevant le placebo.
h La variation moyenne en pourcentage par rapport à la valeur initiale pour la dapagliflozine 10 mg versus placebo, respectivement, était : cholestérol total 2,5 % versus 0,0 % ; HDL cholestérol 6,0 % versus 2,7 % ; LDL cholestérol 2,9 % versus -1,0 % ; triglycérides -2,7 % versus -0,7 %.
i Voir la rubrique Mises en garde spéciales et précautions d'emploi
j L'effet indésirable a été identifié lors de la surveillance en post-commercialisation. Rash inclut les termes préférés suivants, listés par ordre de fréquence dans les études cliniques : rash, rash généralisé, éruption prurigineuse, rash maculeux, rash maculopapuleux, rash pustuleux, rash vésiculeux, et rash érythémateux. Dans les études cliniques contrôlées versus placebo et versus substance active (dapagliflozine, N = 5936, l'ensemble des bras contrôles, N = 3403), la fréquence du rash était similaire pour la dapagliflozine (1,4%) et pour les bras contrôles (1,4 %) respectivement.
k Rapportée dans le cadre de l'étude des effets cardiovasculaires conduite chez des patients atteints de diabète de type 2 (DECLARE). La fréquence est basée sur le taux annuel.
* Rapportés chez ≥ 2 % des sujets et chez ≥ 1 % des sujets avec au moins 3 sujets de plus dans le groupe traité par la dapagliflozine 10 mg par rapport au groupe placebo.
** Rapportés par l'investigateur comme possiblement relié, probablement relié ou relié au traitement de l'étude et rapportés chez ≥ 0,2 % chez des sujets et ≥ 0,1 % chez au moins 3 sujets de plus dans le groupe traité par dapagliflozine 10 mg par rapport au groupe placebo.
Description de certains effets indésirables
Vulvovaginite, balanite et infections génitales associées
Dans l'analyse poolée de 13 études visant à analyser la tolérance, des cas de vulvovaginite, de balanite et d'infections génitales associées ont été rapportés respectivement chez 5,5 % et 0,6 % des patients ayant reçu la dapagliflozine 10 mg et le placebo. La plupart des infections étaient légères à modérées et les patients ont répondu à un traitement standard initial et ont rarement arrêté le traitement par dapagliflozine. Ces infections ont été plus fréquentes chez les femmes (8,4 % et 1,2 % pour la dapagliflozine et le placebo, respectivement) et les patients avec un antécédent étaient plus susceptibles d'avoir une infection récurrente.
Dans l'étude DECLARE, les nombres de patients présentant des événements indésirables graves de type infections génitales étaient faibles et équilibrés : 2 patients dans chacun des groupes dapagliflozine et placebo.
Dans l'étude DAPA-CKD, 3 (0,1 %) patients ont présenté des événements indésirables graves de type infections génitales dans le groupe dapagliflozine et aucun dans le groupe placebo. Trois (0,1 %) patients ont présenté des événements indésirables entraînant l'arrêt du traitement en raison d'infections génitales dans le groupe dapagliflozine et aucun dans le groupe placebo. Aucun événement indésirable grave et aucun arrêt du traitement en raison d'un événement indésirable n'ont été rapportés chez des patients non diabétiques.
Fasciite nécrosante du périnée (gangrène de Fournier)
Des cas de gangrène de Fournier ont été rapportés en post-commercialisation chez des patients prenant des inhibiteurs de SGLT2, incluant la dapagliflozine (voir rubrique Mises en garde et précautions d'emploi).
Dans l'étude DECLARE conduite chez 17 160 patients avec un diabète de type 2 et avec un temps médian d'exposition de 48 mois, un total de 6 cas de gangrène de Fournier ont été rapportés, un dans le groupe traité par la dapagliflozine et 5 dans le groupe placebo.
Hypoglycémie
La fréquence de l'hypoglycémie dépendait du type de traitement initial utilisé dans les études cliniques dans le diabète.
Pour les études de la dapagliflozine en monothérapie, en association à la metformine ou en association à la sitagliptine (avec ou sans metformine), la fréquence des épisodes mineurs d'hypoglycémie s'est avérée similaire (< 5 %) entre les groupes de traitement, y compris le placebo jusqu'à 102 semaines de traitement. Dans toutes les études, les événements majeurs d'hypoglycémie ont été peu fréquents et comparables entre les groupes traités par la dapagliflozine ou le placebo. Les études en association aux sulfamides hypoglycémiants et aux traitements par insuline avaient des taux plus élevés d'hypoglycémie (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Dans une étude en association au glimépiride, aux semaines 24 et 48, des épisodes mineurs d'hypoglycémie ont été rapportés plus fréquemment dans le groupe traité par dapagliflozine 10 mg et glimépiride (6,0 % et 7,9 %, respectivement) que chez les patients ayant reçu le placebo et le glimépiride (2,1 % et 2,1 %, respectivement).
Dans une étude en association à l'insuline, des épisodes d'hypoglycémie majeure ont été rapportés, respectivement aux semaines 24 et 104, chez 0,5 % et 1,0 % du groupe de patients traités par dapagliflozine 10 mg et insuline, et chez 0,5 % du groupe de patients traités par placebo et insuline aux semaines 24 et 104. Aux semaines 24 et 104, des épisodes mineurs d'hypoglycémie ont été rapportés respectivement chez 40,3 % et 53,1 % des patients ayant reçu dapagliflozine 10 mg et insuline et chez 34,0 % et 41,6 % des patients ayant reçu le placebo et insuline.
Dans une étude en association à la metformine et à un sulfamide hypoglycémiant conduite jusqu'à 24 semaines, aucun épisode d'hypoglycémie majeure n'a été rapporté. Des épisodes mineurs d'hypoglycémie ont été rapportés chez 12,8 % des sujets qui ont reçu la dapagliflozine 10 mg plus metformine et un sulfamide hypoglycémiant et chez 3,7 % des sujets qui ont reçu un placebo plus metformine et un sulfamide hypoglycémiant.
Dans l'étude DECLARE, aucune augmentation du risque d'hypoglycémie majeure n'a été observée avec le traitement par dapagliflozine par rapport au placebo. Des événements majeurs d'hypoglycémie ont été rapportés chez 58 (0,7 %) patients traités par dapagliflozine et chez 83 (1,0 %) patients traités par placebo.
Dans l'étude DAPA-CKD, des événements majeurs d'hypoglycémie ont été rapportés chez 14 (0,7 %) patients dans le groupe dapagliflozine et 28 (1,3 %) patients dans le groupe placebo. Ils ont été observés uniquement chez les patients atteints de diabète de type 2.
Déplétion volémique
Dans l'analyse poolée de 13 études visant à analyser la tolérance, des effets évocateurs d'une déplétion volémique (y compris, des cas de déshydratation, d'hypovolémie ou d'hypotension) ont été rapportés chez 1,1 % et 0,7 % des patients ayant reçu respectivement la dapagliflozine 10 mg et le placebo. Des réactions graves sont survenues chez < 0,2 % des patients, et se sont réparties de manière équilibrée entre les patients traités par dapagliflozine 10 mg et le placebo (voir rubrique Mises en garde et précautions d'emploi).
Dans l'étude DECLARE, les nombres de patients présentant des événements évocateurs d'une déplétion volémique étaient équilibrés entre les groupes de traitement : 213 (2,5 %) et 207 (2,4 %) respectivement, dans les groupes dapagliflozine et placebo. Des événements indésirables graves ont été rapportés chez 81 (0,9 %) et 70 (0,8 %) des patients dans les groupes dapagliflozine et placebo, respectivement. Les événements étaient globalement équilibrés entre les groupes de traitement dans les sous-groupes constitués en fonction de l'âge, de l'utilisation de diurétiques, de la pression artérielle et de l'utilisation d'inhibiteurs de l'enzyme de conversion (IEC) / antagonistes des récepteurs de l'angiotensine II (ARA-II). Chez les patients présentant un DFGe <60 mL/min/1,73 m2 à l'inclusion, il y a eu 19 événements indésirables graves évocateurs d'une déplétion volémique dans le groupe dapagliflozine et 13 événements dans le groupe placebo.
Dans l'étude DAPA-HF, le nombre de patients présentant des événements indésirables évocateurs d'une déplétion volémique était de 170 (7,2 %) patients dans le groupe dapagliflozine et 153 (6,5 %) dans le groupe placebo. Moins de patients ont présenté des événements indésirables graves évocateurs d'une déplétion volémique dans le groupe dapagliflozine par rapport au groupe placebo : 23 (1,0 %) et 38 (1,6 %) patients, respectivement. Des résultats similaires ont été observés indépendamment de la présence ou non d'un diabète à l'inclusion et des valeurs initiales du DFGe.
Dans l'étude DAPA-CKD, le nombre de patients présentant des événements indésirables évocateurs d'une déplétion volémique était de 120 (5,6 %) dans le groupe dapagliflozine et de 84 (3,9 %) dans le groupe placebo. Il y a eu 16 (0,7 %) patients avec des événements graves de symptômes évocateurs d'une déplétion volémique dans le groupe dapagliflozine et 15 (0,7 %) patients dans le groupe placebo.
Acidocétose diabétique dans le diabète de type 2
Dans l'étude DECLARE, avec une durée d'exposition médiane de 48 mois, des événements de type ACD ont été rapportés chez 27 patients du groupe dapagliflozine 10 mg et chez 12 patients du groupe placebo. Les événements sont survenus de manière homogène tout au long de la période d'étude. Sur les 27 patients ayant présenté des événements de type ACD dans le groupe dapagliflozine, 22 recevaient également un traitement par insuline au moment de l'événement. Les facteurs déclenchants de l'ACD étaient ceux attendus pour une population de patients atteints de diabète de type 2 (voir rubrique Mises en garde et précautions d'emploi).
Dans l'étude DAPA-HF, des événements de type ACD ont été rapportés chez 3 patients atteints de diabète de type 2 dans le groupe dapagliflozine et aucun dans le groupe placebo.
Dans l'étude DAPA-CKD, des événements de type ACD n'ont été rapportés chez aucun patient dans le groupe dapagliflozine et chez 2 patients atteints de diabète de type 2 dans le groupe placebo.
Dans les deux études cliniques contrôlées versus placebo portant sur la dapagliflozine dans le diabète de type 1, il a été conseillé aux patients de surveiller leur cétonémie en cas de suspicion de symptômes d'ACD et de consulter un médecin si la cétonémie affichée sur leur lecteur était ≥ 0,6 mmol/L. Dans les données poolées à 52 semaines, des événements de type ACD ont été rapportés chez 22 (4,0 %) patients du groupe dapagliflozine 5 mg et 6 (1,1 %) patients du groupe placebo, avec des taux d'incidence correspondants pour 100 patients-années de 4,62 pour la dapagliflozine 5 mg et de 1,27 pour le placebo. Les événements de type ACD sont survenus de manière homogène tout au long de la période d'étude. Des doses inadéquates d'insuline (oubli d'une dose d'insuline ou défaillance de la pompe à insuline) étaient les facteurs déclenchants les plus fréquents. Six événements de type ACD sur 23 dans le groupe dapagliflozine 5 mg sont survenus chez des patients en situation d'euglycémie (< 14 mmol/L ou 250 mg/dL).
Infections des voies urinaires
Dans l'analyse poolée de 13 études visant à analyser la tolérance, les infections des voies urinaires ont été plus fréquemment rapportées chez les patients ayant reçu dapagliflozine 10 mg comparativement au placebo (respectivement, 4,7 % versus 3,5 % ; voir rubrique Mises en garde et précautions d'emploi). La plupart des infections étaient légères à modérées, les patients ont répondu à un traitement standard initial et ont rarement entraîné l'arrêt du traitement par dapagliflozine. Ces infections ont été plus fréquentes chez les femmes, et les patients ayant un antécédent étaient plus susceptibles d'avoir une infection récurrente.
Dans l'étude DECLARE, les événements graves de type infections des voies urinaires ont été rapportés moins fréquemment avec la dapagliflozine 10 mg par rapport au placebo, à savoir 79 (0,9 %) événements versus 109 (1,3 %) événements, respectivement.
Dans l'étude DAPA-HF, le nombre de patients présentant des événements indésirables graves de type infections des voies urinaires était de 14 (0,6 %) patients dans le groupe dapagliflozine et 17 (0,7 %) dans le groupe placebo. Cinq (0,2 %) patients ont présenté des événements indésirables entraînant l'arrêt du traitement en raison d'infections des voies urinaires dans chacun des groupes dapagliflozine et placebo.
Dans l'étude DAPA-CKD, le nombre de patients présentant des événements indésirables graves de type infections des voies urinaires était de 29 (1,3 %) patients dans le groupe dapagliflozine et 18 (0,8 %) dans le groupe placebo. Huit (0,4 %) patients ont présenté des événements indésirables entraînant l'arrêt du traitement en raison d'infections des voies urinaires dans le groupe dapagliflozine et 3 (0,1 %) dans le groupe placebo. Le nombre de patients rapportant des événements indésirables graves ou des arrêts du traitement en raison d'événements indésirables de type infections des voies urinaires parmi les patients non diabétiques était très faible et similaire entre les groupes de traitement (6 [0,9 %] versus 4 [0,6 %] pour les événements indésirables graves ; 1 [0,1 %] versus 0 pour les arrêts du traitement en raison d'événements indésirables).
Augmentation de la créatinine
Les effets indésirables liés à une augmentation de la créatinine ont été regroupés (par ex : diminution de la clairance de la créatinine rénale, altération de la fonction rénale, augmentation de la créatininémie et diminution du débit de filtration glomérulaire). Dans l'analyse poolée de 13 études visant à analyser la tolérance, ce groupe d'effets indésirables a été rapporté respectivement chez 3,2 % des patients recevant la dapagliflozine 10 mg et chez 1,8 % des patients recevant le placebo. Chez les patients avec une fonction rénale normale ou une altération légère de la fonction rénale (valeur initiale du DFGe ≥ 60 mL/min/1,73 m2), ce groupe d'effets indésirables a été rapporté chez 1,3 % des patients recevant la dapagliflozine 10 mg et chez 0,8 % des patients recevant le placebo. Ces réactions ont été plus fréquentes chez les patients avec une valeur initiale du DFGe ≥ 30 et < 60 mL/min/1,73 m2 (18,5 % dapagliflozine 10 mg versus 9,3 % placebo).
Des évaluations complémentaires des patients qui avaient présenté des événements indésirables liés à un trouble rénal ont montré que la plupart des patients avaient des modifications de la créatininémie inférieures ou égales à 0,5 mg/dL par rapport à la valeur initiale. Les augmentations de la créatinine ont été généralement transitoires lors d'un traitement continu ou réversibles après l'arrêt du traitement.
Dans l'étude DECLARE, incluant des patients âgés et des patients présentant une insuffisance rénale (DFGe inférieur à 60 mL/min/1,73 m2), le DFGe a diminué avec le temps dans les deux groupes de traitement. À 1 an, le DFGe moyen était légèrement plus faible, et à 4 ans, le DFGe moyen était légèrement plus élevé dans le groupe dapagliflozine que dans le groupe placebo.
Dans l'étude DAPA-HF, le DFGe a diminué au fil du temps dans le groupe dapagliflozine et le groupe placebo. La diminution initiale du DFGe moyen était de -4,3 mL/min/1,73 m2 dans le groupe dapagliflozine et de -1,1 mL/min/1,73 m2 dans le groupe placebo. À 20 mois, la variation par rapport à la valeur initiale du DFGe était similaire entre les groupes de traitement : -5,3 mL/min/1,73 m2 pour la dapagliflozine et -4,5 mL/min/1,73 m2 pour le placebo.
Dans l'étude DAPA-CKD, le DFGe a diminué au fil du temps dans le groupe dapagliflozine et le groupe placebo. La diminution initiale (J1) du DFGe moyen était de -4,0 mL/min/1,73 m2 dans le groupe dapagliflozine et de -0,8 mL/min/1,73 m2 dans le groupe placebo. À 20 mois, la variation par rapport à la valeur initiale du DFGe était de -7,4 mL/min/1,73 m2 dans le groupe dapagliflozine et de - 8,6 mL/min/1,73 m2 dans le groupe placebo.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé doivent déclarer tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : signalement.social-sante.gouv.fr.
AVANT INSTAURATION DU TRAITEMENT, tenir compte des facteurs pouvant prédisposer à une acidocétose.
SURVEILLER la survenue de symptômes non spécifiques tels que : nausées, vomissements,
anorexie, douleurs abdominales, soif intense, difficulté à respirer, confusion,
fatigue inhabituelle ou somnolence (possibilité d'acidocétose
diabétique).
RECOMMANDER aux patients de consulter un médecin en cas de
symptômes tels qu'une douleur, une sensibilité, un érythème ou une tuméfaction
au niveau de la zone génitale ou périnéale, accompagnés de fièvre ou de
malaises.
En raison de son mécanisme d'action, les patients prenant le
traitement auront un test de glucose urinaire positif.
Interférence avec le test 1,5-anhydroglucitol (1,5-AG)
L'évaluation du contrôle glycémique par le test 1,5-AG n'est pas
recommandée étant donné que les mesures du 1,5-AG sont non fiables pour
l'évaluation du contrôle glycémique chez les patients prenant des
inhibiteurs du SGLT2. L'utilisation de méthodes alternatives pour
l'évaluation du contrôle glycémique est conseillée.
Il n'existe aucune donnée relative à l'utilisation de la dapagliflozine chez la femme enceinte. Des études chez le rat ont révélé une toxicité pour le rein en développement durant la période correspondant aux deuxième et troisième trimestres de la grossesse humaine (voir rubrique Données de sécurité précliniques). Par conséquent, l'utilisation de la dapagliflozine n'est pas recommandée au cours des deuxième et troisième trimestres de grossesse.
Le traitement par la dapagliflozine doit être interrompu dès la découverte de la grossesse.
Allaitement
On ne sait pas si la dapagliflozine et/ou ses métabolites sont excrétés dans le lait humain. Les données pharmacodynamiques/toxicologiques disponibles chez l'animal ont mis en évidence une excrétion de la dapagliflozine/de ses métabolites dans le lait, ainsi que des effets pharmacologiquement induits dans le cadre de l'allaitement (voir rubrique Données de sécurité précliniques). Un risque pour le nouveau né/nourrisson ne peut être exclu. La dapagliflozine ne doit pas être utilisée durant l'allaitement.
Fertilité
L'effet de la dapagliflozine sur la fertilité n'a pas été étudié chez les humains. La dapagliflozine n'a entraîné aucun effet sur la fertilité des rats mâles et femelles, quelle que soit la dose testée.
Diurétiques
La dapagliflozine peut majorer l'effet diurétique des thiazides et des diurétiques de l'anse et peut augmenter le risque de déshydratation et d'hypotension (voir rubrique Mises en garde et précautions d'emploi).
Insuline et sécrétagogues d'insuline
L'insuline et les sécrétagogues d'insuline, comme les sulfamides hypoglycémiants, entraînent une hypoglycémie. Ainsi, une dose plus faible d'insuline ou d'un sécrétagogue d'insuline peut être nécessaire pour réduire le risque d'hypoglycémie lorsqu'ils sont utilisés en association avec la dapagliflozine chez les patients atteints de diabète de type 2 (voir rubriques Posologie et mode d'administration et Effets indésirables).
Interactions pharmacocinétiques
Le métabolisme de la dapagliflozine se fait essentiellement via une réaction de glucuronoconjugaison médiée par l'UDP glucuronosyltransférase 1A9 (UGT1A9).
Lors d'études in vitro, la dapagliflozine n'a ni inhibé les cytochromes P450 (CYP) 1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A4, ni induit les CYP1A2, CYP2B6 ou CYP3A4. Ainsi, la dapagliflozine ne devrait pas modifier la clairance métabolique des médicaments coadministrés et métabolisés par ces enzymes.
Effet des autres médicaments sur la dapagliflozine
Les études d'interaction, principalement à dose unique, menées chez des sujets sains suggèrent que la pharmacocinétique de la dapagliflozine n'est pas modifiée par la metformine, la pioglitazone, la sitagliptine, le glimépiride, le voglibose, l'hydrochlorothiazide, le bumétanide, le valsartan ou la simvastatine.
Suite à la coadministration de la dapagliflozine avec la rifampicine (un inducteur de différents transporteurs actifs et substances métabolisantes), une baisse de 22 % de l'exposition systémique à la dapagliflozine (ASC) a été observée, mais sans effet cliniquement significatif sur l'excrétion urinaire du glucose sur 24 heures. Aucun ajustement posologique n'est recommandé. Aucun effet cliniquement pertinent avec d'autres inducteurs (par exemple la carbamazépine, la phénytoïne, le phénobarbital) n'est attendu.
Suite
à la coadministration de la dapagliflozine avec l'acide méfénamique (un
inhibiteur de UGT1A9), une augmentation de 55 % de l'exposition
systémique de la dapagliflozine a été observée, mais sans effet cliniquement significatif sur l'excrétion urinaire du glucose sur 24 heures.
Aucun ajustement posologique n'est recommandé.
Effet de la dapagliflozine sur les autres médicaments
Lors d'études d'interactions, principalement à dose unique, menées chez des sujets sains, la dapagliflozine n'a pas modifié la pharmacocinétique de la metformine, de la pioglitazone, de la sitagliptine, du glimépiride, de l'hydrochlorothiazide, du bumétanide, du valsartan, de la digoxine (un substrat de la P-gp) ou de la warfarine (S-warfarine, un substrat du CYP2C9), ou les effets anticoagulants de la warfarine mesurés par l'INR. L'association d'une seule dose de dapagliflozine 20 mg et de simvastatine (un substrat du CYP3A4) a entraîné une augmentation de 19 % de l'ASC de la simvastatine et de 31 % de l'ASC de la simvastatine acide. L'augmentation de l'exposition à la simvastatine et à la simvastatine acide n'est pas considérée cliniquement significative.
Interférence avec le test 1,5-anhydroglucitol (1,5-AG)
L'évaluation du contrôle glycémique par le test 1,5-AG n'est pas recommandée étant donné que les mesures du 1,5-AG sont non fiables pour l'évaluation du contrôle glycémique chez les patients prenant des inhibiteurs du SGLT2. L'utilisation de méthodes alternatives pour l'évaluation du contrôle glycémique est conseillée.
Population pédiatrique
Les études d'interaction n'ont été réalisées que chez l'adulte.
Posologie
Maladie rénale chronique
La dose recommandée est 10 mg de dapagliflozine une fois par jour.
Populations particulières
Insuffisance rénale
Aucun ajustement de la dose n'est nécessaire selon l'état de la fonction rénale.
Chez les patients diabétiques, l'efficacité glycémique de la dapagliflozine est réduite lorsque le débit de filtration glomérulaire (DFG) est < 45 mL/min et est vraisemblablement absente chez les patients atteints d'insuffisance rénale sévère. Par conséquent, si le DFG diminue au-dessous de 45 mL/min, un traitement hypoglycémiant supplémentaire doit être envisagé chez les patients diabétiques (voir rubriques Mises en garde et précautions d'emploi, Effets indésirables, Propriétés pharmacodynamiques et Propriétés pharmacocinétiques).
Insuffisance hépatique
Aucun ajustement de la dose n'est nécessaire chez les patients atteints d'insuffisance hépatique légère ou modérée. Les patients atteints d'insuffisance hépatique sévère ne peuvent pas être inclus dans l'ATUc (critère de non éligibilité).
Sujets âgés (≥ 65 ans)
Aucun ajustement de la dose n'est recommandé selon l'âge.
Population pédiatrique
La tolérance et l'efficacité de dapagliflozine chez les enfants âgés de 0 à < 18 ans n'ont pas encore été établies. Aucune donnée n'est disponible.
Mode d'administration
Forxiga peut être pris par voie orale, une fois par jour, à tout moment de la journée, au cours ou en dehors des repas. Les comprimés doivent être avalés entiers.
Durée de conservation :
3 ans
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
La dapagliflozine n'a pas montré de toxicité chez les sujets sains pour des doses orales uniques jusqu'à 500 mg (soit 50 fois la dose maximale recommandée chez l'homme). Ces sujets présentaient des taux de glucose détectables dans l'urine pendant une durée dose dépendante (au moins 5 jours pour la dose de 500 mg), sans cas de déshydratation, d'hypotension ou de déséquilibre électrolytique et sans effet cliniquement significatif sur l'intervalle QTc. L'incidence d'hypoglycémie était similaire au placebo. Lors des études cliniques au cours desquelles des doses quotidiennes jusqu'à 100 mg (soit 10 fois la dose maximale recommandée chez l'homme) étaient administrées pendant 2 semaines à des sujets sains et à des patients diabétiques de type 2, l'incidence d'hypoglycémie était légèrement plus élevée qu'avec le placebo et n'était pas dose dépendante. Le taux d'évènements indésirables incluant la déshydratation ou l'hypotension étaient comparable au placebo, et aucune modification dose dépendante cliniquement significative n'a été observée pour les paramètres biologiques, incluant les électrolytes sériques et les biomarqueurs de la fonction rénale.
En cas de surdosage, un traitement symptomatique adapté doit être administré en fonction de l'état clinique du patient. L'élimination de la dapagliflozine par hémodialyse n'a pas été étudiée.
Classe pharmacothérapeutique : médicaments utilisés dans le traitement du diabète, inhibiteurs du co-transporteur de sodium-glucose de type 2 (SGLT2), code ATC : A10BK01.
Mécanisme d'action
La dapagliflozine est un inhibiteur très puissant (Ki : 0,55 nM), sélectif et réversible du SGLT2.
L'inhibition du SGLT2 par la dapagliflozine réduit la réabsorption du glucose du filtrat glomérulaire dans le tubule rénal proximal avec une réduction concomitante de la réabsorption du sodium favorisant l'excrétion urinaire du glucose et la diurèse osmotique. La dapagliflozine augmente donc l'apport de sodium au niveau du tubule distal, ce qui est supposé augmenter le rétrocontrôle tubulo-glomérulaire et réduire la pression intra-glomérulaire. Ceci, combiné à une diurèse osmotique, entraîne une réduction de la surcharge volumique, une diminution de la pression artérielle et une réduction de la précharge et de la postcharge, et devrait conduire à des effets bénéfiques sur le remodelage cardiaque. Les autres effets comprennent une augmentation de l'hématocrite et une réduction du poids corporel. Les bénéfices cardiaques et rénaux de la dapagliflozine ne dépendent pas uniquement de l'effet hypoglycémiant et ne sont pas limités aux patients diabétiques comme l'ont démontré les études DAPA-HF et DAPA-CKD.
La dapagliflozine améliore la glycémie à jeun et postprandiale en réduisant la réabsorption rénale du glucose et en favorisant ainsi son excrétion urinaire. Cette excrétion du glucose (effet glycosurique) est observée après la première dose, reste effective durant l'intervalle posologique de 24 heures et se poursuit pendant la durée du traitement. La quantité de glucose éliminée par le rein via ce mécanisme dépend de la glycémie et du DFG. De ce fait, chez les sujets ayant une glycémie normale et/ou un DFG faible, la dapagliflozine a une faible propension à provoquer une hypoglycémie car la quantité de glucose filtré est faible et peut être réabsorbée par les transporteurs SGTL1 et SGTL2 débloqués. La dapagliflozine n'affecte pas la production endogène normale du glucose en réponse à l'hypoglycémie. La dapagliflozine agit indépendamment de la sécrétion et de l'action de l'insuline. Une amélioration du modèle d'homéostasie pour l'évaluation de la fonction des cellules bêta (HOMA cellules bêta) a été observée dans les études cliniques avec la dapagliflozine.
Le SGLT2 est exprimé sélectivement dans le rein. La dapagliflozine n'inhibe pas d'autres transporteurs du glucose importants pour le transport du glucose dans les tissus périphériques et est > 1 400 fois plus sélective pour le SGLT2 que pour le SGLT1, le principal transporteur intestinal chargé de l'absorption du glucose.
Effets pharmacodynamiques
Des augmentations de la quantité de glucose excrétée dans l'urine ont été observées chez les sujets sains et chez ceux atteints de diabète de type 2 suite à l'administration de la dapagliflozine. Près de 70 g de glucose ont été excrétés dans l'urine chaque jour (soit 280 kcal/jour) avec une dose quotidienne de 10 mg de la dapagliflozine administrée pendant 12 semaines à des patients atteints de diabète de type 2. Des signes d'excrétion durable du glucose ont été constatés chez des patients atteints de diabète de type 2 et ayant reçu 10 mg/jour de dapagliflozine pendant 2 ans.
Cette excrétion urinaire du glucose associée à la dapagliflozine entraîne également une diurèse osmotique ainsi qu'une augmentation du volume urinaire chez les patients présentant un diabète de type 2. L'augmentation du volume urinaire chez les patients atteints de diabète de type 2 traités par dapagliflozine 10 mg était maintenue à 12 semaines et s'élevait à environ 375 mL/jour. L'augmentation du volume urinaire était associée à une augmentation légère et transitoire de l'excrétion urinaire du sodium, elle même non liée à une évolution de la concentration de sodium sérique.
L'excrétion urinaire de l'acide urique a également augmenté de manière transitoire (pendant 3 à 7 jours) et a été accompagnée d'une diminution durable de la concentration sérique d'acide urique. A 24 semaines, la diminution de la concentration sérique d'acide urique était comprise entre 48,3 et 18,3 micromoles/L (de 0,87 à 0,33 mg/dL).
Efficacité et sécurité clinique
Pression artérielle
Dans une analyse poolée dans la population de patients diabétiques prédéfinie de 13 études versus placebo, le traitement par la dapagliflozine 10 mg a entraîné une variation de la pression artérielle systolique de 3,7 mmHg et de la pression artérielle diastolique de 1,8 mmHg par rapport aux valeurs initiales, versus 0,5 mmHg pour la pression systolique et 0,5 mmHg pour la pression diastolique dans le groupe placebo à la semaine 24. Des diminutions similaires ont été observées jusqu'à 104 semaines.
Dans deux études contrôlées versus placebo de 12 semaines, un total de 1 062 patients diabétiques de type 2 insuffisamment contrôlés et hypertendus (malgré la prise d'un traitement antérieur régulier par inhibiteur de l'enzyme de conversion de l'angiotensine (IEC) ou par antagoniste des récepteurs de l'angiotensine II (ARA-II) dans une étude et par IEC ou par ARA-II associé à un traitement antihypertenseur additionnel dans l'autre étude) ont été traités par 10 mg de dapagliflozine ou par placebo. Pour les 2 études, à la semaine 12, la dapagliflozine 10 mg associée au traitement antidiabétique habituel a entraîné une amélioration de l'HbA1c et une diminution de la pression artérielle systolique, corrigée par rapport à la diminution due à la prise d'un placebo, respectivement de 3,1 et 4,3 mmHg en moyenne.
Dans une étude spécifique chez des patients diabétiques ayant un DFGe ≥ 45 à < 60 mL/min/1,73 m2, un traitement par dapagliflozine a montré une réduction de la pression artérielle systolique en position assise à la semaine 24 de -4,8 mmHg par rapport à une réduction de 1,7 mmHg pour le placebo (p < 0,05).
Maladie rénale chronique
DAPA-CKD (The Study to Evaluate the Effect of Dapagliflozin on Renal Outcomes and Cardiovascular Mortality in Patients with Chronic Kidney Disease) était une étude internationale, multicentrique, randomisée, en double aveugle, contrôlée versus placebo chez des patients atteints de maladie rénale chronique (MRC) avec un DFGe ≥ 25 à ≤ 75 mL/min/1,73 m2 et une albuminurie (rapport albuminurie/créatininurie [RAC] ≥ 200 et ≤ 5000 mg/g) afin de déterminer l'effet de la dapagliflozine par rapport au placebo, lorsque le médicament était ajouté aux traitements standards en cours, sur l'incidence du critère composite « diminution prolongée du DFGe ≥ 50 %, insuffisance rénale terminale (IRT) (définie comme un DFGe prolongé < 15 mL/min/1,73 m2, une dialyse chronique ou une transplantation rénale), décès cardiovasculaire ou rénal ».
Sur 4 304 patients, 2 152 ont été randomisés pour recevoir la dapagliflozine 10 mg et 2 152 le placebo, et ont été suivis pendant une durée médiane de 28,5 mois. Le traitement était poursuivi si le DFGe diminuait à moins de 25 mL/min/1,73 m2 pendant l'étude et pouvait être poursuivi si une dialyse était nécessaire.
L'âge moyen des patients dans l'étude était de 61,8 ans, 66,9 % étaient des hommes. À l'inclusion, le DFGe moyen était de 43,1 mL/min/1,73 m2 et le RAC médian de 949,3 mg/g, 44,1 % des patients avaient un DFGe 30 à < 45 mL/min/1,73 m2 et 14,5 % un DFGe < 30 mL/min/1,73 m2. 67,5 % des patients avaient un diabète de type 2. Les patients recevaient un traitement standard ; 97,0 % des patients étaient traités par un inhibiteur de l'enzyme de conversion de l'angiotensine (IEC) ou par antagoniste des récepteurs de l'angiotensine (ARA).
La dapagliflozine a été supérieure au placebo concernant la prévention pour le critère composite principal « diminution prolongée du DFGe ≥ 50 %, insuffisance rénale terminale, décès cardiovasculaire ou rénal » (HR 0,61 [IC à 95 % 0,51 ; 0,72] ; p < 0,0001). Le nombre nécessaire à traiter pendant 27 mois était de 19 (IC à 95 % 15 ; 27). D'après l'analyse de Kaplan-Meier, les courbes d'événements pour la dapagliflozine et le placebo ont commencé à se séparer tôt (4 mois) et elles ont continué à diverger sur la période d'étude (Figure 1).
Figure 1 : Délai de survenue du premier événement du critère composite « diminution prolongée du DFGe ≥ 50 %, insuffisance rénale terminale, décès cardiovasculaire ou rénal »
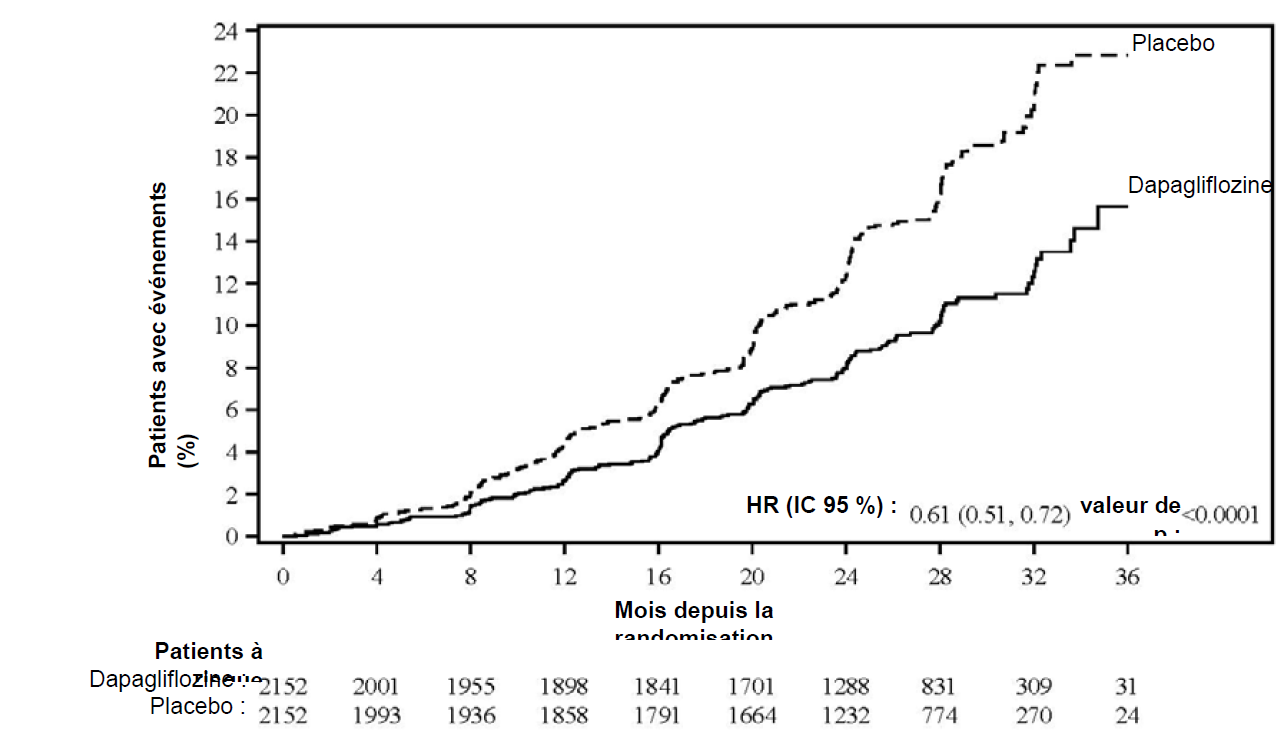
« Patients à risque » correspond au nombre de patients à risque au début de la période.
Les quatre composantes du critère d'évaluation principal composite ont chacune contribué à l'effet du traitement. La dapagliflozine a également réduit l'incidence du critère composite « diminution prolongée du DFGe ≥ 50 %, insuffisance rénale terminale ou décès rénal » (HR 0,56 [IC à 95 % 0,45 ; 0,68], p < 0,0001) et du critère composite « décès cardiovasculaire et hospitalisation pour insuffisance cardiaque » (HR 0,71 [IC à 95 % 0,55 ; 0,92], p = 0,0089) (Figure 2).
Figure 2 : Effets du traitement concernant les critères d'évaluation principal et secondaires composites, leurs composantes et la mortalité toutes causes confondues
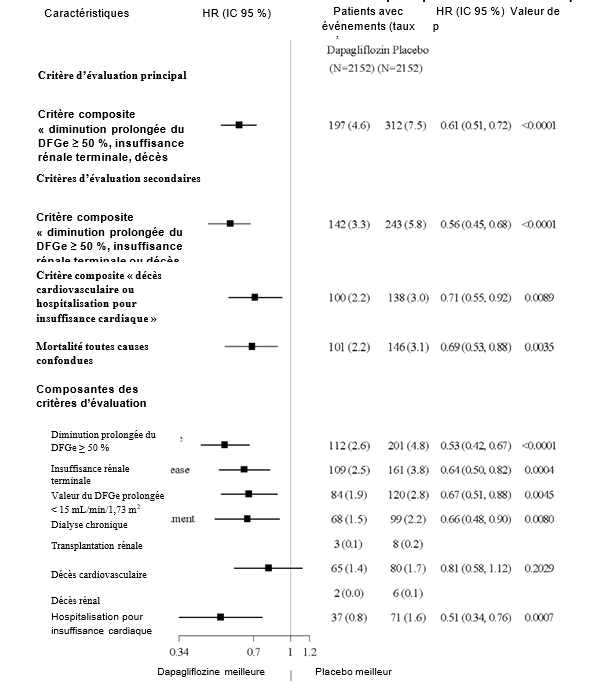
Le nombre de premiers événements pour les composantes individuelles correspond au nombre effectif de premiers événements pour chaque composante et ne s'additionne pas au nombre d'événements du critère d'évaluation composite.
Les taux d'événements sont présentés comme le nombre de patients avec événements pour 100 patients- années de suivi.
Les valeurs de p pour les composantes des critères d'évaluation composites sont nominales.
Le traitement par la dapagliflozine a amélioré la survie globale chez les patients atteints de maladie rénale chronique, avec une réduction significative de la mortalité toutes causes confondues (HR 0,69 [IC à 95 % 0,53 ; 0,88], p = 0,0035) (Figure 3).
Figure 3 : Délai de survenue du décès quelle qu'en soit la cause
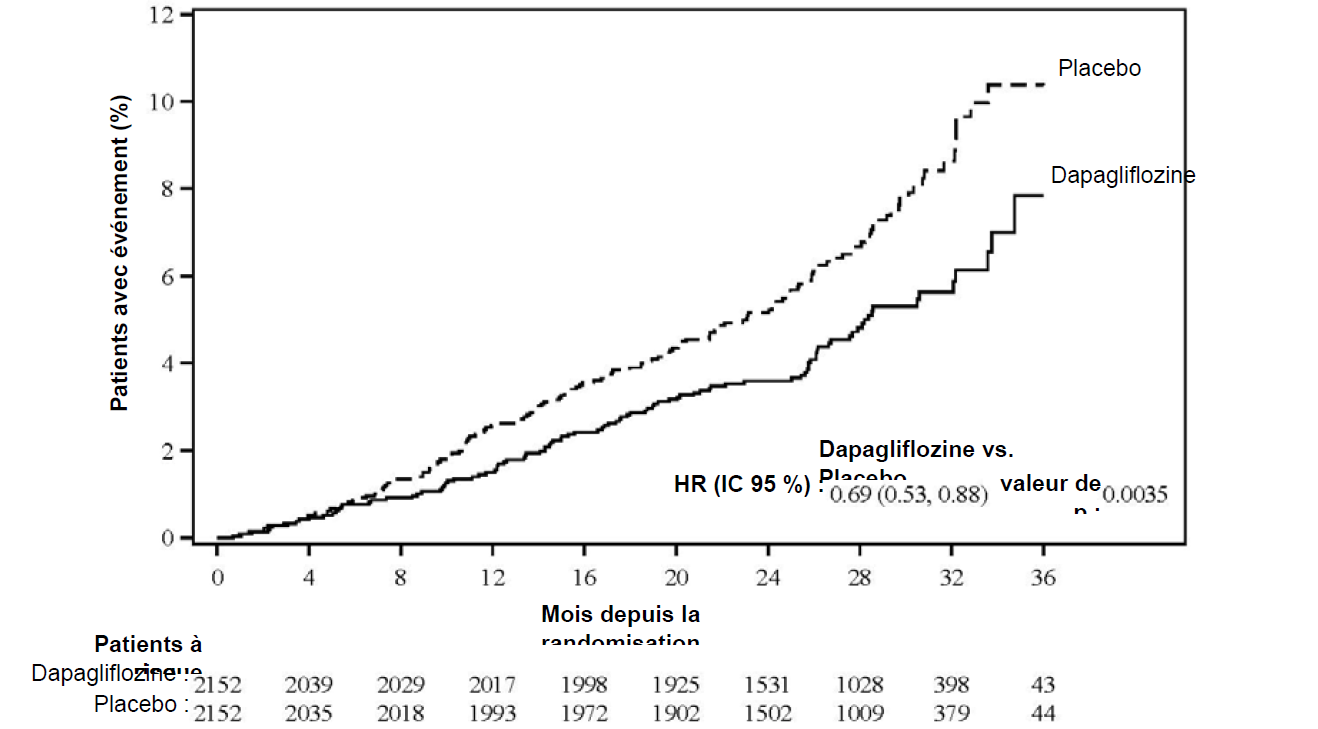
« Patients à risque » correspond au nombre de patients à risque au début de la période.
L'effet du traitement par la dapagliflozine a été cohérent chez les patients atteints de maladie rénale chronique avec un diabète de type 2 ou sans diabète (Figure 4).
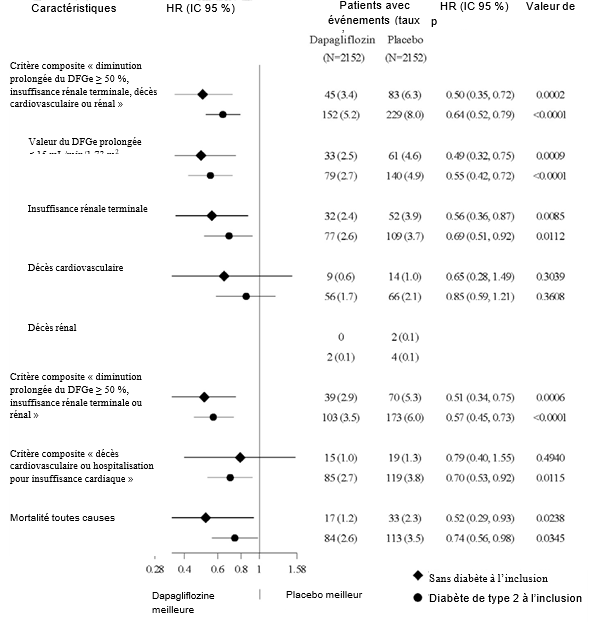
Le
nombre de premiers événements pour les composantes individuelles
correspond au nombre effectif de premiers événements pour chaque
composante et ne s'additionne pas au nombre d'événements du critère
d'évaluation composite.décès
Les estimations du hazard ratio ne sont pas présentées pour les sous-groupes avec moins de 15 événements au total, les deux bras sont regroupés.
Les taux d'événements sont présentés comme le nombre de patients avec événements pour 100 patients- années de suivi.
Les valeurs de p sont nominales.
Le traitement par la dapagliflozine a entraîné une réduction plus importante de l'albuminurie. L'effet a été observé dès 14 jours et s'est maintenu pendant toute la durée de l'étude. À 36 mois, la variation moyenne ajustée en pourcentage par rapport à la valeur initiale du RAC (mg/g) était de -41 % chez les patients traités par la dapagliflozine et de -20 % chez les patients traités par placebo, avec une différence entre les groupes de traitement de -26,3 % ([IC à 95 % -36,8 ; -14,0], valeur de p nominale = 0,0001).
L'incidence de doublement de la créatininémie par rapport à la mesure en laboratoire la plus récente (qui est une évaluation de l'aggravation aiguë de la fonction rénale) a été réduite dans le groupe dapagliflozine par rapport au groupe placebo (HR 0,68 [IC à 95 % 0,49 ; 0,94], p nominale = 0,0187).
Moins de patients ont présenté des événements rénaux dans le groupe dapagliflozine par rapport au groupe placebo : 144 (6,7 %) et 169 (7,9 %) patients, respectivement.
Population pédiatrique
L'Agence Européenne des Médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec la dapagliflozine pour le traitement du diabète de type 2 au sein d'un ou plusieurs sous-groupes de la population pédiatrique (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec la dapagliflozine dans tous les sous-groupes de la population pédiatrique dans la prévention des événements cardiovasculaires chez les patients atteints d'insuffisance cardiaque chronique et dans le traitement de la maladie rénale chronique (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Absorption
La dapagliflozine est rapidement et bien absorbée après administration orale. Les concentrations plasmatiques maximales de dapagliflozine (Cmax) sont généralement atteintes dans les 2 heures suivant la prise à jeun. Les moyennes géométriques des valeurs Cmax et ASCτ à l'état d'équilibre avec une dose quotidienne de 10 mg de dapagliflozine ont été respectivement de 158 ng/mL et 628 ng/mL. La biodisponibilité orale absolue de la dapagliflozine après administration d'une dose de 10 mg atteint 78 %. L'administration avec un repas à forte teneur en graisses a réduit la valeur Cmax de la dapagliflozine jusqu'à 50 % et prolonge la valeur Tmax d'environ 1 heure, sans toutefois modifier l'ASC par rapport à une prise à jeun. Ces changements ne sont pas considérés comme cliniquement significatifs. Forxiga peut donc être administré indifféremment au cours ou en dehors des repas.
Distribution
La dapagliflozine est liée à environ 91 % aux protéines. La liaison protéique n'a pas été modifiée dans diverses conditions pathologiques (par exemple, insuffisance rénale ou hépatique). Le volume moyen de distribution de la dapagliflozine à l'état d'équilibre était de 118 litres.
Biotransformation
La dapagliflozine est largement métabolisée, principalement sous forme de 3 O glucuronide de dapagliflozine, un métabolite inactif. Le 3 O glucuronide de dapagliflozine ou les autres métabolites ne contribuent pas aux effets hypoglycémiants. La formation du 3 O glucuronide de dapagliflozine est médiée par l'UGT1A9, une enzyme présente dans le foie et les reins. Le métabolisme médié par le CYP était considéré comme une voie de clairance mineure chez l'homme.
Élimination
La demi vie plasmatique terminale moyenne (t1/2) de la dapagliflozine est de 12,9 heures après la prise par voie orale d'une seule dose de dapagliflozine 10 mg chez les sujets sains. La clairance systémique totale moyenne de dapagliflozine administrée par voie intraveineuse est de 207 mL/min. La dapagliflozine et les métabolites associés sont principalement éliminés via l'excrétion urinaire, avec moins de 2 % de dapagliflozine sous sa forme inchangée. Après administration d'une dose de 50 mg de [14C] dapagliflozine, 96 % ont été retrouvés, 75 % dans l'urine et 21 % dans les selles. Dans les selles, 15 % environ de la dose est éliminée sous forme de molécule initiale.
Linéarité
L'exposition à la dapagliflozine augmente proportionnellement à la dose allant de 0,1 à 500 mg. Des administrations quotidiennes répétées jusqu'à 24 semaines n'ont pas modifié sa pharmacocinétique dans le temps.
Populations spécifiques
Insuffisance rénale
À l'état d'équilibre (dose quotidienne unique de 20 mg de la dapagliflozine pendant 7 jours), les patients atteints d'un diabète de type 2 et d'une insuffisance rénale légère, modérée ou sévère (déterminée par la clairance plasmatique de l'iohexol) présentaient une exposition systémique moyenne à la dapagliflozine supérieure de 32 %, 60 % et 87 %, respectivement, à celle des diabétiques de type 2 ayant une fonction rénale normale. L'excrétion urinaire du glucose à 24 heures à l'état d'équilibre dépendait fortement de la fonction rénale. Les patients atteints d'un diabète de type 2 et présentant une fonction rénale normale ou une insuffisance rénale légère, modérée ou sévère ont ainsi respectivement éliminé 85, 52, 18 et 11 g de glucose/jour. L'impact de l'hémodialyse sur l'exposition à la dapagliflozine n'est pas connu. L'effet de la réduction de la fonction rénale sur l'exposition systémique a été évalué dans une modèle de pharmacocinétique de population. En accord avec les résultats précédents, l'ASC prédite par le modèle a été plus élevée chez les patients atteints de maladie rénale chronique que chez les patients ayant une fonction rénale normale et elle n'a pas été significativement différente chez les patients atteints de maladie rénale chronique avec un diabète de type 2 ou sans diabète.
Insuffisance hépatique
Chez les patients atteints d'insuffisance hépatique légère ou modérée, (classe Child Pugh A et B), les valeurs Cmax et ASC moyennes de la dapagliflozine étaient respectivement supérieures de 12 % et 36 %, à celles des témoins appariés sains. Ces différences n'ont pas été considérées comme cliniquement significatives. Chez les patients atteints d'insuffisance hépatique sévère (classe Child Pugh C), les valeurs Cmax et ASC moyennes de la dapagliflozine étaient supérieures de 40 % et 67 %, respectivement, à celles des témoins sains.
Sujets âgés (≥ 65 ans)
Aucune augmentation cliniquement significative de l'exposition en fonction de l'âge seul n'a été mise en évidence chez les patients jusqu'à 70 ans. Toutefois, une exposition accrue due à la détérioration de la fonction rénale liée à l'âge peut être attendue. Il n'existe pas de données suffisantes pour conclure sur l'exposition des patients de plus de 70 ans.
Population pédiatrique
La pharmacocinétique dans la population pédiatrique n'a pas été étudiée.
Sexe
L'ASCee moyenne de la dapagliflozine chez les femmes est estimée supérieure de 22 % environ à celle des hommes.
Origine ethnique
Aucune différence cliniquement significative n'a été observée entre les patients d'origine caucasienne, d'origine afro américaine ou d'origine asiatique en matière d'exposition systémique.
Poids corporel
L'exposition à la dapagliflozine était diminuée par la prise de poids. En conséquence, les patients de faible poids peuvent avoir une exposition plus ou moins augmentée et les patients avec un poids élevé peuvent avoir une exposition plus ou moins diminuée. Toutefois, les différences d'exposition n'ont pas été considérées comme cliniquement significatives.
Forxiga n'a pas d'effet ou qu'un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines. Les patients doivent être informés du risque d'hypoglycémie lorsque la dapagliflozine est administrée en association avec des sulfamides hypoglycémiants ou de l'insuline.
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration répétée, génotoxicité, cancérogénèse, et des fonctions de reproduction et de développement, n'ont pas révélé de risque particulier pour l'homme. La dapagliflozine n'a pas induit de tumeurs que ce soit chez les souris ou les rats à chacune des doses évaluées lors des études de carcinogénicité d'une durée de deux ans.
Toxicité pour les fonctions de reproduction et de développement
L'administration directe de dapagliflozine à de jeunes rats en période de sevrage et l'exposition indirecte en fin de gestation (périodes correspondant aux deuxième et troisième trimestres de grossesse en matière de maturation rénale humaine) et pendant l'allaitement sont chacune associées à une incidence et/ou une sévérité accrue des dilatations rénales pelviennes et tubulaires chez la descendance.
Dans une étude de toxicité juvénile, de jeunes rats ont fait l'objet d'une administration directe de dapagliflozine entre le jour postnatal 21 et le jour postnatal 90. Des dilatations rénales pelviennes et tubulaires ont été rapportées à tous les niveaux de dose. L'exposition des petits à la plus faible dose testée était ≥ 15 fois la dose maximale recommandée chez l'homme. Ces résultats étaient associés à une augmentation dose dépendante du poids des reins et à une hypertrophie macroscopique des reins observée à toutes les doses. Les dilatations rénales pelviennes et tubulaires constatées chez les jeunes animaux n'ont pas été totalement réversibles au cours de la période de récupération d'environ 1 mois.
Lors d'une étude distincte du développement pré et postnatal, des rates ont été traitées à compter du jour 6 de gestation jusqu'au jour postnatal 21. Il en résulte que les petits ont été exposés indirectement in utero et tout au long de l'allaitement. (Une étude satellite a été menée pour évaluer l'exposition à la dapagliflozine dans le lait et chez les petits). Une incidence ou une sévérité accrue des dilatations rénales pelviennes chez les descendants adultes des mères traitées a été observée, mais uniquement à la plus forte dose testée (les expositions correspondantes des mères et des petits à la dapagliflozine étaient, respectivement, 1 415 fois et 137 fois supérieures aux valeurs obtenues à la dose maximale recommandée chez l'homme). La toxicité additionnelle pour les fonctions de développement était limitée à une perte de poids dose dépendante chez les petits et n'a été observée qu'à des doses ≥ 15 mg/kg/jour. Elle était associée à une exposition ≥ 29 fois, par rapport aux valeurs obtenues à la dose maximale recommandée chez l'homme. La toxicité maternelle était uniquement évidente à la plus forte dose testée et se limitait à des pertes de poids et d'appétit temporaires. La dose sans effet nocif observé (NOEL) concernant la toxicité pour les fonctions de développement, autrement dit la plus faible dose testée, était associée à une exposition maternelle systémique environ 19 fois supérieure à la valeur obtenue à la dose maximale recommandée chez l'homme.
Lors d'études complémentaires portant sur le développement embryo foetal chez les rats et les lapins, la dapagliflozine a été administrée selon des intervalles coïncidant avec les phases majeures de l'organogénèse de chacune des espèces. Aucune des doses testées n'a induit de toxicité maternelle ou sur le développement chez les lapins. La plus forte dose testée était associée à une exposition systémique environ 1 191 fois supérieure à la dose maximale recommandée chez l'homme. Chez les rats, la dapagliflozine ne s'est révélée ni embryolétale ni tératogène à des expositions jusqu'à 1 441 fois supérieures à la dose maximale recommandée chez l'homme.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Médicament soumis à prescription hospitalière.
Médicament à prescription réservée aux néphrologues.
Comprimé pelliculé (comprimé).
Comprimés pelliculés, jaunes, biconvexes, en forme de losange, d'approximativement 1,1 x 0,8 cm de diagonale, avec « 10 » gravé sur une face et « 1428 » gravé sur l'autre face.
Plaquette thermoformée alu/alu
Présentation de 30 x 1 comprimés pelliculés dans des plaquettes thermoformées unitaires perforées.
Dapagliflozine.................................................................................................................. 10 mg
Excipient(s) à effet notoire : Chaque comprimé contient 50 mg de lactose. Pour la liste complète des excipients, voir rubrique Liste des excipients.
Noyau du comprimé
Cellulose microcristalline (E460i)
Lactose
Crospovidone (E1202)
Dioxyde de silicium (E551)
Stéarate de magnésium (E470b)
Pelliculage
Alcool polyvinylique (E1203)
Dioxyde de titane (E171)
Macrogol 3350 (E1521)
Talc (E553b)
Oxyde de fer jaune (E172)