
OPDIVO 10 mg-mL, solution à diluer pour perfusion, boîte de 1 flacon de 4 ml
Retiré du marché le : 26/11/2021
Dernière révision : 02/04/2021
Taux de TVA : 0%
Laboratoire exploitant : BRISTOL-MYERS SQUIBB
Source :

Mésothéliome Pleural Malin
OPDIVO est indiqué en association à l'ipilimumab, en première ligne, dans le traitement des patients adultes atteints d'un mésothéliome pleural malin non résécable.
Hypersensibilité au principe actif ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Effets indésirables d'origine immunologique
Lorsque nivolumab est administré en association à l'ipilimumab, se référer au Résumé des Caractéristiques du Produit d'ipilimumab avant l'initiation du traitement. Les effets indésirables d'origine immunologique sont survenus à des fréquences plus élevées lorsque nivolumab était administré en association à l'ipilimumab comparativement à nivolumab en monothérapie. La plupart des effets indésirables d'origine immunologique s'est améliorée ou résolue avec une prise en charge appropriée, incluant l'initiation de corticoïdes et des modifications de traitement (voir rubrique Posologie et mode d'administration).
Des effets indésirables cardiaques et pulmonaires, notamment des embolies pulmonaires ont aussi été rapportés avec le traitement en association. Les patients doivent être continuellement surveillés pour des effets indésirables cardiaques et pulmonaires, ainsi que pour des signes cliniques, des symptômes et des anomalies biologiques indiquant des troubles électrolytiques et une déshydratation, avant l'initiation et à intervalles réguliers au cours du traitement.
Nivolumab en association à l'ipilimumab doit être arrêté en cas d'effets indésirables sévères cardiaques et pulmonaires récurrents ou pouvant menacer le pronostic vital (voir rubrique Posologie et mode d'administration).Les patients doivent être continuellement surveillés (au moins jusqu'à 5 mois après la dernière perfusion), un effet indésirable avec nivolumab ou avec nivolumab en association à l'ipilimumab pouvant survenir à tout moment pendant ou après l'arrêt du traitement.
En cas de suspicion d'effets indésirables d'origine immunologique, une évaluation appropriée doit être effectuée afin de confirmer l'étiologie ou d'exclure d'autres causes. Sur la base de la sévérité de l'effet indésirable, le traitement par nivolumab ou par nivolumab en association à l'ipilimumab doit être suspendu, et des corticoïdes administrés. Si une immunosuppression par corticoïdes est utilisée pour traiter un effet indésirable, une décroissance progressive des doses sur une période d'au moins un mois doit être initiée à partir de l'amélioration. Une diminution rapide des doses peut entraîner une aggravation ou une récidive de l'effet indésirable. Des traitements immunosuppresseurs non stéroïdiens doivent être ajoutés en cas d'aggravation ou d'absence d'amélioration malgré l'utilisation de corticoïdes.
Le traitement par nivolumab ou par nivolumab en association à l'ipilimumab ne doit pas être repris tant que le patient reçoit des doses immunosuppressives de corticoïdes ou d'autres médicaments immunosuppresseurs. Une prophylaxie antibiotique doit être utilisée pour prévenir les infections opportunistes chez les patients recevant des médicaments immunosuppresseurs.
Nivolumab ou nivolumab en association à l'ipilimumab doit être définitivement arrêté en cas d'effet indésirable sévère récurrent d'origine immunologique, et pour tout effet indésirable d'origine immunologique pouvant menacer le pronostic vital.
Pneumopathie inflammatoire d'origine immunologique
Des pneumopathies inflammatoires ou interstitielles sévères, dont des cas d'issue fatale, ont été observées avec nivolumab en monothérapie ou nivolumab en association à l'ipilimumab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour déceler l'apparition de signes et symptômes de pneumopathie inflammatoire, tels que des modifications radiologiques (ex : opacités focales en verre dépoli, infiltrats localisés), dyspnée et hypoxie. Une étiologie infectieuse et toute autre pathologie liée à la maladie doivent être éliminées.
En cas de pneumopathie inflammatoire de Grade 3 ou 4, nivolumab ou nivolumab en association à l'ipilimumab doit être arrêté définitivement et une corticothérapie à la dose de 2 à 4 mg/kg/jour de méthylprednisolone ou équivalent doit être débutée.
En cas de pneumopathie inflammatoire (symptomatique) de Grade 2, nivolumab ou nivolumab en association à l'ipilimumab doit être suspendu et une corticothérapie à la dose de 1 mg/kg/jour de méthylprednisolone ou équivalent doit être débutée. Après amélioration, nivolumab ou nivolumab en association à l'ipilimumab peut être repris après réduction progressive des corticoïdes. En cas d'aggravation ou d'absence d'amélioration malgré l'initiation d'une corticothérapie, les doses de corticoïdes doivent être augmentées à la dose de 2 à 4 mg/kg/jour de méthylprednisolone ou équivalent, et nivolumab ou nivolumab en association à l'ipilimumab doit être arrêté définitivement.
Colite d'origine immunologique
Des diarrhées ou des colites sévères ont été observées avec nivolumab en monothérapie ou nivolumab en association à l'ipilimumab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour déceler l'apparition de diarrhées et d'autres symptômes de colites, tels que des douleurs abdominales et la présence de mucus ou de sang dans les selles. Une infection / réactivation du cytomégalovirus (CMV) a été rapportée chez des patients ayant des colites d'origine immunologique réfractaires aux corticoïdes. Les étiologies infectieuses et autres des diarrhées doivent être exclues, par conséquent, des tests de biologie appropriés et des examens complémentaires doivent être effectués. Si le diagnostic de colite d'origine immunologique réfractaire aux corticoïdes est confirmé, l'ajout d'un agent immunosuppresseur alternatif au traitement par corticoïdes ou le remplacement du traitement par corticoïdes doivent être envisagés.
En cas de diarrhée ou de colite de Grade 4, nivolumab ou nivolumab en association à l'ipilimumab doit être arrêté définitivement et une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit être débutée.
Nivolumab en monothérapie doit être suspendu en cas de diarrhée ou de colite de Grade 3, et une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit être débutée. Après amélioration, nivolumab en monothérapie peut être repris après réduction progressive des corticoïdes. En cas d'aggravation ou d'absence d'amélioration malgré l'initiation de corticoïdes, nivolumab en monothérapie doit être définitivement arrêté.
L'observation de diarrhée ou colite de Grade 3 avec nivolumab en association à l'ipilimumab nécessite aussi un arrêt définitif du traitement et l'initiation de corticoïdes à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent.
En cas de diarrhée ou de colite de Grade 2, nivolumab ou nivolumab en association à l'ipilimumab doit être suspendu. Les diarrhées ou les colites persistantes doivent être traitées par une corticothérapie à la dose de 0,5 à 1 mg/kg/jour de méthylprednisolone ou équivalent. Après amélioration, nivolumab ou nivolumab en association à l'ipilimumab peut être repris après réduction progressive des corticoïdes, si nécessaire. En cas d'aggravation ou d'absence d'amélioration malgré l'initiation d'une corticothérapie, les doses de corticoïdes doivent être augmentées à 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent, et nivolumab ou nivolumab en association à l'ipilimumab doit être arrêté définitivement.
Hépatite d'origine immunologique
Des hépatites sévères ont été observées avec nivolumab en monothérapie ou nivolumab en association à l'ipilimumab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour déceler l'apparition de signes et symptômes d'hépatite, tels que des augmentations des transaminases et de la bilirubine totale. Une étiologie infectieuse et toute autre pathologie liée à la maladie doivent être éliminées.
En cas d'élévation de Grade 3 ou 4 des transaminases ou de la bilirubine totale, nivolumab ou nivolumab en association à l'ipilimumab doit être arrêté définitivement et une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit être débutée.
En cas d'élévation de Grade 2 des transaminases ou de la bilirubine totale, nivolumab ou nivolumab en association à l'ipilimumab doit être suspendu. La persistance de cette élévation des valeurs biologiques doit être prise en charge par une corticothérapie à la dose de 0,5 à 1 mg/kg/jour de méthylprednisolone ou équivalent. Après amélioration, nivolumab ou nivolumab en association à l'ipilimumab peut être repris après réduction progressive des corticoïdes, si nécessaire. En cas d'aggravation ou d'absence d'amélioration malgré l'initiation d'une corticothérapie, les doses de corticoïdes doivent être augmentées à 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent et nivolumab ou nivolumab en association à l'ipilimumab doit être arrêté définitivement.
Néphrite et dysfonction rénale d'origine immunologique
Des néphrites et des atteintes rénales sévères ont été observées avec le traitement en monothérapie ou nivolumab en association à l'ipilimumab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour déceler l'apparition de signes et symptômes de néphrite ou d'atteinte rénale. La plupart des patients ont présenté des augmentations asymptomatiques de la créatinine sérique. Toute autre étiologie liée à la maladie doit être écartée.
En cas d'élévation de Grade 4 de la créatinine sérique, nivolumab ou nivolumab en association à l'ipilimumab doit être arrêté définitivement et une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit être débutée.
En cas d'élévation de Grade 2 ou 3 de la créatinine sérique, nivolumab ou nivolumab en association à l'ipilimumab doit être suspendu et une corticothérapie à la dose de 0,5 à 1 mg/kg/jour de méthylprednisolone ou équivalent doit être débutée. Après amélioration, nivolumab ou nivolumab en association à l'ipilimumab peut être repris après réduction progressive des corticoïdes. En cas d'aggravation ou d'absence d'amélioration malgré la corticothérapie, les doses de corticoïdes doivent être augmentées à 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent, et nivolumab ou nivolumab en associationà l'ipilimumab doit être arrêté définitivement.
Endocrinopathies d'origine immunologique
Des endocrinopathies sévères, incluant hypothyroïdie, hyperthyroïdie, insuffisance surrénalienne (incluant l'insuffisance cortico-surrénalienne secondaire), hypophysite (incluant l'hypopituitarisme), diabète, et acidocétose diabétique ont été observées avec nivolumab en monothérapie ou nivolumab en association à l'ipilimumab (voir rubrique Effets indésirables).
Les patients doivent être surveillés pour déceler l'apparition de signes et symptômes d'endocrinopathie, et d'hyperglycémie et des modifications de la fonction thyroïdienne (au début du traitement, à intervalles réguliers en cours de traitement, et si cliniquement indiqué).
En cas d'hypothyroïdie symptomatique, nivolumab ou nivolumab en association à l'ipilimumab doit être suspendu, et un traitement substitutif en hormone thyroïdienne doit être débuté, si nécessaire. En cas d'hyperthyroïdie symptomatique, nivolumab ou nivolumab en association à l'ipilimumab doit être suspendu, et un traitement par antithyroïdiens doit être débuté, si nécessaire. Une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit également être envisagée, en cas de suspicion d'une inflammation aiguë de la thyroïde. Après amélioration, nivolumab ou nivolumab en association à l'ipilimumab peut être repris après réduction progressive des corticoïdes, si nécessaire. La surveillance de la fonction thyroïdienne doit être poursuivie afin de s'assurer que le traitement substitutif hormonal approprié est utilisé. Nivolumab ou nivolumab en association à l'ipilimumab doit être arrêté définitivement en cas d'hyperthyroïdie ou d'hypothyroïdie pouvant menacer le pronostic vital.
En cas d'insuffisance surrénalienne symptomatique de Grade 2, nivolumab ou nivolumab en association à l'ipilimumab doit être suspendu, et une corticothérapie substitutive à une dose physiologique doit être débutée, si nécessaire. Nivolumab ou nivolumab en association à l'ipilimumab doit être arrêté définitivement en cas d'insuffisance surrénalienne sévère (Grade 3) ou pouvant menacer le pronostic vital (Grade 4). La surveillance de la fonction surrénalienne et des taux d'hormone doit être poursuivie afin de s'assurer que la corticothérapie substitutive appropriée est utilisée.
En cas d'hypophysite symptomatique de Grade 2 ou 3, nivolumab ou nivolumab en association à l'ipilimumab doit être suspendu, et un traitement substitutif hormonal doit être débuté, si nécessaire. En cas de suspicion d'inflammation aiguë de la glande pituitaire, une corticothérapie à la dose de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent doit également être envisagée. Après amélioration, nivolumab ou nivolumab en association à l'ipilimumab peut être repris après réduction progressive des corticoïdes, si nécessaire. Nivolumab ou nivolumab en association à l'ipilimumab doit être arrêté définitivement en cas d'hypophysite pouvant menacer le pronostic vital (Grade 4). La surveillance de la fonction pituitaire et des taux d'hormone doit être poursuivie afin de s'assurer que le traitement substitutif hormonal approprié est utilisé.
En cas de diabète symptomatique, nivolumab ou nivolumab en association à l'ipilimumab doit être suspendu, et un traitement substitutif par insuline doit être débuté, si nécessaire. La surveillance de la glycémie doit être poursuivie afin d'assurer que le traitement substitutif par insuline approprié est utilisé. Nivolumab ou nivolumab en association à l'ipilimumab doit être arrêté définitivement en cas de diabète pouvant menacer le pronostic vital.
Effets indésirables cutanés d'origine immunologique
Des éruptions cutanées sévères ont été observées avec nivolumab en association à l'ipilimumab et, moins fréquemment, avec nivolumab en monothérapie (voir rubrique Effets indésirables). Nivolumab ou nivolumab en association à l'ipilimumab doit être suspendu en cas d'éruption cutanée de Grade 3, et il doit être arrêté définitivement en cas d'éruption cutanée de Grade 4. Les éruptions cutanées sévères doivent être prises en charge avec de hautes doses de corticoïdes, de 1 à 2 mg/kg/jour de méthylprednisolone ou équivalent.
De rares cas de SJS et de NET, dont certains d'issue fatale, ont été observés. En cas d'apparition de signes ou symptômes de SJS ou de NET, le traitement par nivolumab ou par nivolumab en association à l'ipilimumab doit être interrompu et le patient adressé à un service spécialisé pour évaluation et traitement. Si le patient a développé un SJS ou une NET lors de l'utilisation de nivolumab ou de nivolumab en association à l'ipilimumab, l'arrêt définitif du traitement est recommandé (voir rubrique Posologie et mode d'administration).
L'utilisation de nivolumab doit être considérée avec précaution chez un patient ayant présenté un effet indésirable cutané sévère ou ayant menacé le pronostic vital lors d'un précédent traitement anticancéreux stimulant l'immunité.
Autres effets indésirables d'origine immunologique
Les effets indésirables d'origine immunologique suivants ont été rapportés chez moins de 1% des patients traités par nivolumab en monothérapie ou par nivolumab en association à l'ipilimumab dans les essais cliniques avec différentes doses et dans différents types de tumeurs : pancréatite, uvéite, démyélinisation, neuropathie auto-immune (incluant parésie des nerfs facial et abducens), syndrome de Guillain-Barré, myasthénie grave, syndrome myasthénique, méningite aseptique, encéphalite, gastrite, sarcoïdose, duodénite, myosite, myocardite et rhabdomyolyse. Des cas de syndrome de Vogt-Koyanagi-Harada et d'hypoparathyroïdie ont été rapportés après commercialisation (voir rubrique Effets indésirables).
En cas de suspicion d'effet indésirable d'origine immunologique, une évaluation appropriée doit être effectuée afin de confirmer l'étiologie ou d'exclure d'autres causes. En fonction de la gravité de l'effet indésirable, le traitement par nivolumab ou par nivolumab en association à l'ipilimumab doit être suspendu et des corticoïdes doivent être administrés. Après amélioration, nivolumab ou nivolumab en association à l'ipilimumab peut être repris après réduction progressive des corticoïdes. Nivolumab ou nivolumab en association à l'ipilimumab doit être arrêté définitivement en cas d'effet indésirable sévère récurrent d'origine immunologique, et pour tout effet indésirable d'origine immunologique pouvant menacer le pronostic vital.
Des cas de myotoxicité (myosite, myocardite et rhabdomyolyse), dont certains d'issue fatale, ont été rapportés avec nivolumab ou nivolumab en association à l'ipilimumab. Si un patient développe des signes et symptômes de myotoxicité, une surveillance étroite doit être mise en place et le patient doit être adressé à un spécialiste pour évaluation et traitement sans délai. Sur la base de la sévérité de la myotoxicité, nivolumab ou nivolumab en association à l'ipilimumab doit être suspendu ou arrêté (voir rubrique Posologie et mode d'administration), et un traitement approprié instauré.
Le diagnostic de myocardite exige un haut degré de suspicion. Les patients présentant des symptômes cardiaques ou cardio-pulmonaires doivent être évalués pour une myocardite potentielle. Si une myocardite est suspectée, l'administration immédiate d'une dose élevée de stéroïdes (prednisone 1 à 2 mg/kg/jour ou méthylprednisolone 1 à 2 mg/kg/jour) et une consultation immédiate en cardiologie avec un bilan diagnostique, selon les directives cliniques actuelles, doivent être initiées. Une fois qu'un diagnostic de myocardite est établi, le traitement par nivolumab ou par nivolumab en association à l'ipilimumab doit être suspendu ou définitivement interrompu (voir rubrique Posologie et mode d'administration).
Un rejet de greffe d'organe solide a été signalé après la mise sur le marché chez des patients traités par inhibiteurs du PD-1. Le traitement par nivolumab peut augmenter le risque de rejet chez les bénéficiaires d'une greffe d'organe solide. Il convient de prendre en considération le rapport entre les bénéfices du traitement par nivolumab et le risque de rejet d'organe chez ces patients.
Des cas de lymphohistiocytose hémophagocytaire (LHH) ont été observés avec le nivolumab en monothérapie et en association à l'ipilimumab. L'administration de nivolumab en monothérapie ou en association à l'ipilimumab doit être faite avec précaution. Si une LHH est confirmée, l'administration de nivolumab en monothérapie ou en association à l'ipilimumab doit être interrompue et un traitement contre la LHH doit être instauré.
Réactions à la perfusion
Des réactions sévères liées à la perfusion ont été rapportées dans les essais cliniques de nivolumab ou de nivolumab en association à l'ipilimumab (voir rubrique Effets indésirables). En cas de réaction sévère à la perfusion ou pouvant menacer le pronostic vital, la perfusion de nivolumab ou de nivolumab en association à l'ipilimumab doit être arrêtée et un traitement médical approprié doit être administré. Les patients présentant une réaction à la perfusion d'intensité légère à modérée peuvent recevoir nivolumab ou nivolumab en association à l'ipilimumab sous surveillance étroite et avec l'utilisation d'une prémédication suivant les recommandations locales de traitement pour la prophylaxie des réactions liées à la perfusion.
Précautions spécifiques à la maladie
Mésothéliome pleural malin
Les patients présentant un mésothéliome primitif péritonéal, péricardique, ou de la tunique vaginale des testicules, une maladie pulmonaire interstitielle, une maladie auto-immune active, toute maladie nécessitant une immunosuppression systémique, et des métastases cérébrales (à moins d'une résection chirurgicale ou d'une radiothérapie stéréotaxique, et sans évolution dans les 3 mois précédant l'inclusion dans l'étude) ont été exclus de l'étude pivot dans le traitement de première ligne du MPM (voir rubriques Interactions avec d'autres médicaments et autres formes d'interactions et Propriétés pharmacodynamiques).
En l'absence de données, nivolumab en association à l'ipilimumab doit être utilisé avec précaution dans ces populations, après évaluation attentive du bénéfice/risque potentiel au cas par cas.
Patients sous régime hyposodé contrôlé
Chaque mL de ce médicament contient 0,1 mmol (ou 2,5 mg) de sodium. Ce médicament contient 10 mg de sodium par flacon de 4 ml, 25 mg de sodium par flacon de 10 ml ou 60 mg de sodium par flacon de 24 ml, ce qui équivaut à 0,5 %, 1,25 % ou 3 % respectivement, de l'apport quotidien maximal recommandé par l'OMS de 2 g de sodium pour un adulte.
Carte d'alerte patient
Tous les prescripteurs d'OPDIVO doivent connaître l'information destinée aux médecins et les recommandations de prise en charge. Le prescripteur doit discuter avec le patient des risques liés au traitement par OPDIVO. Le patient recevra à chaque prescription la carte d'alerte patient.
Nivolumab en monothérapie (voir rubrique Posologie et mode d'administration)
Résumé du profil de sécurité
Dans l'ensemble des données poolées de nivolumab en monothérapie dans différents types de tumeur (n = 2787) avec un suivi minimum de 2,3 à 28 mois, les effets indésirables les plus fréquents (≥ 10% des patients) ont été : fatigue (29%), éruption cutanée (17%), prurit (13%), diarrhées (13%), et nausées (12%). La plupart des effets indésirables était d'intensité légère à modérée
(Grade 1 ou 2). Avec un suivi minimum de 63 mois dans le CBNPC, aucun nouveau signal relatif à la sécurité n'a été identifié.
Liste tabulée des effets indésirables
Les effets indésirables rapportés dans l'ensemble des données poolées pour les patients traités par nivolumab en monothérapie (n = 2787) sont présentés dans le Tableau 2. Ces effets sont présentés par classe de systèmes d'organes et par ordre de fréquence. Les fréquences sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1000 à < 1/100), rare (≥ 1/10000 à < 1/1000), très rare (< 1/10000) ; fréquence indéterminée (ne peut être estimée sur la base des données post-commercialisation disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre de gravité décroissant.
Tableau 2 : Effets indésirables du nivolumab en monothérapie
| | Nivolumab en monothérapie |
| Infections et infestations | |
| Fréquent | infection des voies aériennes supérieures |
| Peu Fréquent | pneumoniea, bronchite |
| Fréquence indéterminée | méningite aseptiqueh |
| Tumeurs bénignes, malignes et non précisées (y compris kystes et polypes) | |
| Rare | lymphadénite histiocytique nécrosante (lymphadénite de Kikuchi) |
| Affections hématologiques et du système lymphatique | |
| Très fréquent | neutropéniea,b |
| Peu fréquent | éosinophilie |
| Fréquence indéterminée | lymphohistiocytose hémophagocytaire |
| Affections du système immunitaire | |
| Fréquent | réaction liée à la perfusionc, hypersensibilitéc |
| Rare | réaction anaphylactiquec |
| Fréquence indéterminée | rejet de greffe d'organe solideh, sarcoïdoseh |
| Affections endocriniennes | |
| Fréquent | hypothyroïdie, hyperthyroïdie |
| Peu fréquent | insuffisance surrénaliennel, hypopituitarisme, hypophysite, thyroïdite, diabète sucré |
| Rare | acidocétose diabétique |
| Fréquence indéterminée | hypoparathyroïdieh |
| Troubles du métabolisme et de la nutrition | |
| Fréquent | diminution de l'appétit |
| Peu fréquent | déshydratation, acidose métabolique |
| Fréquence indéterminée | syndrome de lyse tumoralei |
| Affections du système nerveux | |
| Fréquent | neuropathie périphérique, céphalée, sensation vertigineuse |
| Peu fréquent | polyneuropathie, neuropathie auto-immune (incluant parésie des nerfs facial et abducens) |
| Rare | syndrome de Guillain-Barré, démyélinisation, syndrome myasthénique, encéphalitea,c,m |
| Affections oculaires | |
| Peu fréquent | uvéite, vision trouble, sécheresse oculaire |
| Fréquence indéterminée | Syndrome de Vogt-Koyanagi-Haradah |
| Affections cardiaques | |
| Peu fréquent | tachycardie, affections du péricardej |
| Rare | arythmie (incluant arythmie ventriculaire)d, fibrillation auriculaire, myocarditea, f |
| Affections vasculaires | |
| Fréquent | hypertension |
| Rare | vascularite |
| Affections respiratoires, thoraciques et médiastinales | |
| Fréquent | pneumopathie inflammatoirea,c, dyspnéea, toux |
| Peu fréquent | épanchement pleural |
| Rare | infiltration pulmonaire |
| Affections gastro-intestinales | |
| Très fréquent | diarrhée, nausée |
| Fréquent | colitea, stomatite, vomissement, douleur abdominale, constipation, sécheresse buccale |
| Peu fréquent | pancréatite, gastrite |
| Rare | ulcère duodénal |
| Affections hépatobiliaires | |
| Peu fréquent | hépatitec |
| Rare | cholestase |
| Affections de la peau et du tissu sous-cutané | |
| Très fréquent | éruption cutanéee, prurit |
| Fréquent | vitiligo, peau sèche, érythème, alopécie |
| Peu fréquent | érythème polymorphe, psoriasis, rosacée, urticaire |
| Rare | nécrolyse épidermique toxiquea,f, syndrome de Stevens-Johnsona, f |
| Affections musculo-squelettiques et systémiques | |
| Fréquent | douleur musculo-squelettiqueg, arthralgie |
| Peu fréquent | pseudopolyarthrite rhizomélique, arthrite |
| Rare | syndrome de Sjögren, myopathie, myosite (notamment polymyosite)a,f, rhabdomyolysea,f |
| Affections du rein et des voies urinaires | |
| Peu fréquent | néphrite tubulo-interstitielle, insuffisance rénale (incluant atteinte rénale aiguë)a,c |
| Troubles généraux et anomalies au site d'administration | |
| Très fréquent | fatigue |
| Fréquent | fièvre, oedème (incluant oedème périphérique) |
| Peu fréquent | douleur, douleur thoracique |
| Investigationsb | |
| Très fréquent | augmentation du taux d'ASAT, augmentation du taux d'ALAT, augmentation du taux de phosphatases alcalines, augmentation de la lipase, augmentation de l'amylase, hypocalcémie, augmentation du taux de créatinine, hyperglycémiec, hypoglycémie, lymphopénie, leucopénie, thrombocytopénie, anémiek, hypercalcémie, hyperkaliémie, hypokaliémie, hypomagnésémie, hyponatrémie |
| Fréquent | augmentation du taux de bilirubine totale, hypermagnésémie, hypernatrémie, perte de poids |
a Des cas d'issue fatale ont été rapportés dans les études cliniques terminées ou en cours.
b Les fréquences représentent la proportion de patients ayant présenté une aggravation des valeurs biologiques par rapport aux valeurs à l'inclusion. Voir ci-dessous: « Description des effets indésirables sélectionnés ; anomalies des valeurs biologiques ».
c Des cas pouvant menacer le pronostic vital ont été rapportés dans les études cliniques terminées ou en cours.
d Dans la population avec un mélanome métastatique prétraitée par CTLA4/BRAF inhibiteur, la fréquence des effets indésirables dans le système classe-organe des affections cardiaques, indépendamment de la causalité, était plus élevée dans le groupe nivolumab que dans le groupe chimiothérapie. Les taux d'incidence pour 100 patients-années d'exposition étaient de 9,3 vs 0; des événements cardiaques graves ont été rapportés chez 4,9% des patients dans le groupe nivolumab vs 0 dans le groupe de traitement selon le choix de l'investigateur. Dans la population avec un mélanome métastatique non prétraitée, la fréquence des effets indésirables cardiaques était plus basse dans le groupe nivolumab que dans le groupe dacarbazine. Tous ces effets indésirables ont été considérés par les investigateurs comme non liés au traitement par nivolumab, à l'exception des arythmies (fibrillation auriculaire, tachycardie et arythmie ventriculaire).
e Eruption cutanée est un terme composite incluant éruption cutanée maculopapuleuse, éruption cutanée érythémateuse, éruption cutanée prurigineuse, éruption cutanée folliculaire, éruption cutanée maculaire, éruption cutanée morbiliforme, éruption cutanée papuleuse, éruption cutanée pustuleuse, éruption cutanée papulosquameuse, éruption cutanée vésiculaire, éruption cutanée généralisée, éruption cutanée exfoliative, dermatite, dermatite acnéiforme, dermatite allergique, dermatite atopique, dermatite bulleuse, dermatite exfoliative, dermatite psoriasiforme, toxidermie et pemphigoïde.
f Rapporté également dans des études en dehors des données poolées. La fréquence est basée sur l'exposition dans l'ensemble du programme.
g Douleur musculosquelettique est un terme composite qui inclut douleur dorsale, douleur osseuse, douleur de type musculosquelettique dans la poitrine, inconfort musculosquelettique, myalgie, douleur du cou, douleur des extrémités et douleur spinale.
h Evènement rapporté post-commercialisation (voir également rubrique Mises en garde et précautions d'emploi).
i Rapporté dans les études cliniques et dans le cadre de la post-commercialisation.
j Affections du péricarde est un terme composite incluant péricardite, épanchement péricardique, tamponnade cardiaque, et syndrome de Dressler.
k Anémie est un terme composite incluant, parmi d'autres causes, l'anémie hémolytique et l'anémie auto- immune.
l Inclut insuffisance surrénalienne et insuffisance corticosurrénalienne secondaire
m Inclut encéphalite et encéphalite limbique
Nivolumab en association à l'ipilimumab (voir rubrique Posologie et mode d'administration)
Résumé du profil de sécurité
Lorsque nivolumab est administré en association à l'ipilimumab, consulter le Résumé des Caractéristiques du Produit d'ipilimumab avant d'initier le traitement. Pour de plus amples informations sur le profil de tolérance d'ipilimumab en monothérapie, veuillez consulter le RCP d'ipilimumab.
MPM
Dans l'ensemble des données de nivolumab 3 mg/kg en association à l'ipilimumab 1 mg/kg dans le MPM (n = 300), avec un suivi minimum de 17,4 mois, les effets indésirables les plus fréquents
(≥ 10%) ont été : fatigue (43%), diarrhée (31%), éruption cutanée (30%), douleur musculosquelettique (27%), nausée (24%), diminution de l'appétit (24%), prurit (21%), constipation (19%) et hypothyroïdie (13%). La plupart des effets indésirables était d'intensité légère à modérée (Grade 1 ou 2).
Liste tabulée des effets indésirables
Les effets indésirables rapportés dans l'ensemble des données poolées chez les patients traités par nivolumab 3 mg/kg en association à l'ipilimumab 1 mg/kg dans le MPM (n = 300) sont présentés dans le Tableau 3. Ces effets sont présentés par classe de systèmes d'organes et par ordre de fréquence. Les fréquences sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données post-commercialisation disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre de gravité décroissant.
Tableau 3: Effets indésirables du nivolumab en association à l'ipilimuab
| | Nivolumab 3 mg/kg en association à l'ipilimumab 1 mg/kg dans le MPM*** | |
| Affections du système immunitaire | ||
| Fréquent | réaction liée à la perfusion, hypersensibilité | |
| Affections endocriniennes | ||
| Très fréquent | hypothyroïdie | |
| Fréquent | hyperthyroïdie, insuffisance surrénalienne, hypophysite, hypopituitarisme | |
| Peu fréquent | thyroïdite | |
| Troubles du métabolisme et de la nutrition | ||
| Très fréquent | diminution de l'appétit | |
| Affections du système nerveux | ||
| Peu fréquent | encéphalite | |
| Affections cardiaques | ||
| Peu fréquent | myocardite | |
| Affections respiratoires, thoraciques et médiastinales | ||
| Fréquent | pneumopathie inflammatoire | |
| Affections gastro-intestinales | ||
| Très fréquent | diarrhée, nausée, constipation | |
| Fréquent | colite, pancréatite | |
| Affections hépatobiliaires | ||
| Fréquent | hépatite | |
| Affections de la peau et du tissu sous-cutané | ||
| Très fréquent | éruption cutanéeb, prurit | |
| Affections musculo-squelettiques et systémiques | ||
| Très fréquent | douleur musculo-squelettiquec | |
| Fréquent | arthrite | |
| Peu fréquent | myosite | |
| Affections du rein et des voies urinaires | |
| Fréquent | atteinte rénale aiguë, insuffisance rénale |
| Troubles généraux et anomalies au site d'administration | |
| Très fréquent | fatigue |
| Investigationsb | |
| Très fréquent | augmentation du taux d'ASAT, augmentation du taux d'ALAT, augmentation du taux de phosphatases alcalines, augmentation de la lipase, augmentation de l'amylase, augmentation du taux de créatinine, hyperglycémiea, lymphopénie, anémied, hypercalcémie, hypocalcémie, hyperkaliémie, hypokaliémie, hyponatrémie, hypomagnésémie |
| Fréquent | augmentation du taux de bilirubine totale, hypoglycémie, leucopénie, neutropéniea, thrombocytopénie, hypernatrémie, hypermagnésémie |
![]()
![]()
![]()
![]() *** nivolumab en association à l'ipilimumab dans le MPM.
*** nivolumab en association à l'ipilimumab dans le MPM.
a Des cas d'issue fatale ont été rapportés dans les études cliniques terminées ou en cours.
b Eruption cutanée est un terme composite incluant éruption cutanée macropapuleuse, éruption cutanée érythémateuse, éruption cutanée prurigineuse, éruption cutanée folliculaire, éruption cutanée maculaire, éruption cutanée morbiliforme, éruption cutanée papuleuse, éruption cutanée pustuleuse, éruption cutanée papulosquameuse, éruption cutanée vésiculaire, éruption cutanée généralisée, éruption cutanée exfoliative, dermatite, dermatite acnéiforme, dermatite allergique, dermatite atopique, dermatite bulleuse, dermatite exfoliative, dermatite psoriasiforme, toxidermie et pemphigoïde.
c Douleur musculosquelettique est un terme composite qui inclut douleur dorsale, douleur osseuse, douleur de type musculosquelettique dans la poitrine, inconfort musculosquelettique, myalgie, douleur du cou, douleur des extrémités et douleur spinale.
d Anémie est un terme composite incluant, parmi d'autres causes, l'anémie hémolytique et l'anémie auto-immune.
Description des effets indésirables sélectionnés
Nivolumab ou nivolumab en association à l'ipilimumab est associé à des effets indésirables d'origine immunologique. Avec un traitement médical approprié, les effets indésirables d'origine immunologique ont été résolus dans la plupart des cas. L'arrêt définitif du traitement a été nécessaire chez une plus grande proportion de patients recevant nivolumab en association à l'ipilimumab que chez ceux recevant nivolumab en monothérapie. Le tableau 4 présente le pourcentage des patients ayant développé des effets indésirables d'origine immunologique qui ont définitivement arrêté le traitement, en fonction du schéma posologique. En outre, chez les patients qui ont présenté un événement, le tableau 4 présente le pourcentage des patients qui ont nécessité des doses élevées de corticoïdes (au moins 40 mg de prednisone ou équivalent par jour), en fonction du schéma posologique. Les recommandations de prise en charge de ces effets indésirables sont décrites en rubrique Mises en garde et précautions d'emploi.
Tableau 4: Effets indésirables d'origine immunologique entraînant l'arrêt définitif ou nécessitant des doses élevées de corticoïdes, en fonction du schéma posologique ((nivolumab en monothérapie ou en association avec l'ipilimumab)
| | Nivolumab 3 mg/kg ou 240 mg en monothérapie % | Nivolumab 3 mg/kg en association à l'ipilimumab 1 mg/kg dans le MPM % |
| Effet indésirable d'origine immunologique entraînant l'arrêt définitif | | |
| Pneumopathie inflammatoire | 1,4 | 2,3 |
| Colite | 0,8 | 5,0 |
| Hépatite | 1,0 | 3,7 |
| Néphrite et dysfonctionnement rénal | 0,3 | 1,3 |
| Endocrinopathies | 0,2 | 0,3 |
| Peau | 0,4 | 0,7 |
| Hypersensibilité/Réaction à la perfusion | 0,2 | 1,7 |
| Effet indésirable d'origine immunologique nécessitant des doses élevées de corticoïdesa,b | | |
| Pneumopathie inflammatoire | 67 | 70 |
| Colite | 14 | 33 |
| Hépatite | 19 | 42 |
| Néphrite et dysfonctionnement rénal | 26 | 40 |
| Endocrinopathies | 7 | 10 |
| Peau | 4 | 8 |
| Hypersensibilité/Réaction à la perfusion | 20 | 17 |
a Au moins 40 mg d'équivalent prednisone par jour
b La fréquence est basée sur le nombre de patients qui ont présenté l'effet indésirable d'origine immunologique.
Pneumopathie inflammatoire d'origine immunologique
Chez les patients traités par nivolumab en monothérapie, l'incidence des pneumopathies inflammatoires, incluant pneumopathie interstitielle et infiltration pulmonaire, était de 3,6% (99/2787). La majorité des cas étaient de Grade 1 ou 2 en sévérité et ont été rapportés chez 0,9% (24/2787) et 1,8% (51/2787) des patients, respectivement. Des cas de Grade 3 et 4 ont été rapportés chez 0,8% (21/2787) et <0,1% (1/2787) des patients, respectivement. Des cas de Grade 5 ont été rapportés chez <0,1% (2/2787) des patients dans ces études. Le délai médian de survenue était de 3,3 mois (de 0,2 à 19,6 mois). La résolution est survenue chez 66 patients (66,7%) avec un délai médian de résolution de 6,6 semaines (de 0,1+ à 96,7+ semaines) ; + indique une donnée censurée.
Chez les patients traités par nivolumab 3 mg/kg en association à l'ipilimumab 1 mg/kg dans le MPM, l'incidence des pneumopathies inflammatoires, incluant des pneumopathies interstitielles, était de 6,7% (20/300). Des cas de Grade 2 et Grade 3 ont été rapportés chez respectivement 5,3% (16/300) et 0,7% (2/300) des patients. Le délai médian de survenue était de 1,8 mois (de 0,3 à 20,8 mois). Une résolution est survenue chez 16 patients (80%) avec un délai médian de résolution de 6,1 semaines (de 1,1 à 113,1+ semaines).
Colite d'origine immunologique
Chez les patients traités par nivolumab en monothérapie, l'incidence des diarrhées, colites ou selles fréquentes était de 13% (361/2787). La majorité des cas étaient de Grade 1 ou 2 en sévérité et ont été rapportés chez 8,3% (230/2787) et 3,2% (88/2787) des patients, respectivement. Des cas de Grade 3 ont été rapportés chez 1,5% (43/2787) des patients. Aucun cas de Grade 4 ou 5 n'a été rapporté dans ces études. Le délai médian de survenue était de 1,8 mois (de 0,0 à 26,6 mois). Une résolution est survenue chez 311 patients (86,9%) avec un délai médian de résolution de 2,1 semaines (de 0,1 à 124,4+ semaines).
Chez les patients traités par nivolumab 3 mg/kg en association à l'ipilimumab 1 mg/kg dans le MPM, l'incidence des diarrhées ou colites était de 22,0% (66/300). Des cas de Grade 2 et Grade 3 ont été rapportés chez respectivement 7,3% (22/300) et 5,3% (16/300) des patients. Le délai médian de survenue était de 3,9 mois (de 0,0 à 21,7 mois). Une résolution est survenue chez 62 patients (93,9%) avec un délai médian de résolution de 3,1 semaines (de 0,1 à 100,0+ semaines).
Hépatite d'origine immunologique
Chez les patients traités par nivolumab en monothérapie, l'incidence des anomalies de la fonction hépatique était de 6,7% (187/2787). La majorité des cas étaient de Grade 1 ou 2 en sévérité et ont été rapportés chez 3,5% (98/2787) et 1,4% (38/2787) des patients respectivement. Des cas de Grade 3 et 4 ont été rapportés chez 1,5% (42/2787) et 0,3% (9/2787) des patients, respectivement. Aucun cas de Grade 5 n'a été rapporté dans ces études. Le délai médian de survenue était de 2,1 mois (de 0,0 à 27,6 mois). Une résolution est survenue chez 142 patients (76,3%) avec un délai médian de résolution de 5,9 semaines (de 0,1 à 94,3+ semaines).
Chez les patients traités par nivolumab 3 mg/kg en association à l'ipilimumab 1 mg/kg dans le MPM, l'incidence des anomalies de la fonction hépatique était de 12,0% (36/300). Des cas de Grade 2, Grade 3 et Grade 4 ont été rapportés chez respectivement 1,7% (5/300), 4,3% (13/300) et 1,0% (3/300) des patients. Le délai médian de survenue était de 1,8 mois (de 0,5 à 20,3 mois). Une résolution est survenue chez 31 patients (86,1%) avec un délai médian de résolution de 4,1 semaines (de 1,0 à 78,3+ semaines).
Néphrite et dysfonction rénale d'origine immunologique
et 4 ont été rapportés chez 0,4% (12/2787) et <0,1% (1/2787) des patients, respectivement. Aucun cas de néphrite ou de dysfonction rénale de Grade 5 n'a été rapporté dans ces études. Le délai médian de survenue était de 2,3 mois (de 0,0 à 18,2 mois). Une résolution est survenue chez 45 patients (63,4%) avec un délai médian de résolution de 12,1 semaines (de 0,3 à 79,1+ semaines).
Chez les patients traités par nivolumab 3 mg/kg en association à l'ipilimumab 1 mg/kg dans le MPM, l'incidence des dysfonctions rénales était de 5,0% (15/300). Des cas de Grade 2 et de Grade 3 ont été rapportés chez respectivement 2,0% (6/300) et 1,3% (4/300) des patients. Le délai médian de survenue était de 3,6 mois (de 0,5 à 14,4). Une résolution est survenue chez 12 patients (80,0%) avec un délai médian de résolution de 6,1 semaines (de 0,9 à 126,4+).
Endocrinopathies d'origine immunologique
Chez les patients traités par nivolumab en monothérapie, l'incidence des troubles thyroïdiens, incluant hypothyroïdie et hyperthyroïdie, était de 9,7% (270/2787). La majorité des cas étaient de Grade 1 ou 2 en sévérité et ont été rapportés chez 4,2% (118/2787) et 5,4% (150/2787) des patients, respectivement. Des troubles thyroïdiens de Grade 3 ont été rapportés chez <0,1% (2/2787) des patients. Des hypophysites (1 de Grade 1, 2 de Grade 2, 5 de Grade 3 et 1 de Grade 4), des hypopituitarismes (5 de Grade 2 et 1 de Grade 3), des insuffisances surrénaliennes (incluant une insuffisance corticosurrénalienne secondaire) (1 de Grade 1, 9 de Grade 2, et 5 de Grade 3), des cas de diabète (incluant un diabète de type 1) (3 de Grade 2 et 1 de Grade 3) et des acidocétoses diabétiques (2 de Grade 3) ont été rapportés. Aucun cas de Grade 5 n'a été rapporté dans ces études. Le délai médian de survenue de ces endocrinopathies était de 2,8 mois (de 0,3 à 29,1 mois). Une résolution est survenue chez 123 patients (41,6%). Le délai de résolution allait de 0,4 à 144,1+ semaines.
Chez les patients traités par nivolumab 3 mg/kg en association à l'ipilimumab 1 mg/kg dans le MPM, l'incidence des troubles thyroïdiens était de 14% (43/300). Des troubles thyroïdiens de Grade 2 et de Grade 3 ont été rapportés chez respectivement 9,3% (28/300) et 1,3% (4/300) des patients. Des hypophysites sont survenues chez 2% (6/300) des patients. Des cas de Grade 2 ont été rapportés chez 1,3% (4/300) des patients. Des hypopituitarismes de Grade 2 et de Grade 3 sont survenus chez respectivement 1,0% (3/300) et 1,0% (3/300) des patients. Des insuffisances surrénaliennes de Grade 2 et Grade 3 sont survenues chez respectivement 1,7% (5/300) et 0,3% (1/300) des patients. Le délai médian de survenue de ces endocrinopathies était de 2,8 mois (de 0,5 à 20,8 mois). Une résolution est survenue chez 17 patients (32,7%). Le délai de résolution allait de 0,3 à 144,1+ semaines.
Effets indésirables cutanés d'origine immunologique
Chez les patients traités par nivolumab en monothérapie, l'incidence des éruptions cutanées était de 25,9% (722/2787). La majorité des cas étaient de Grade 1 en sévérité et ont été rapportés chez 19,6% (546/2787) des patients. Des cas de Grade 2 et de Grade 3 ont été rapportés chez 5,0% (139/2787) et 1,3% (37/2787) des patients, respectivement. Aucun cas de Grade 4 ou 5 n'a été rapporté dans ces études. Le délai médian de survenue était de 1,4 mois (de 0,0 à 27,9 mois). Une résolution est survenue chez 448 patients (62,8%), avec un délai médian de résolution de 17,4 semaines (de 0,1 à 150,0+ semaines).
Chez les patients traités par nivolumab 3 mg/kg en association à l'ipilimumab 1 mg/kg dans le MPM, l'incidence des éruptions cutanées était de 36,0% (108/300). Des cas de Grade 2 et Grade 3 ont été rapportés chez respectivement 10,3% (31/300) et 3,0% (9/300) des patients. Le délai médian de survenue était de 1,6 mois (de 0,0 à 22,3 mois). Une résolution est survenue chez 71 patients (66,4%) avec un délai médian de résolution de 12,1 semaines (de 0,4 à 146,4+ semaines).
De rares cas de SJS et de NET dont certains d'issue fatale ont été observés (voir rubriques Posologie et mode d'administration et Mises en garde et précautions d'emploi).
Réactions à la perfusion
Chez les patients traités par nivolumab en monothérapie, l'incidence des hypersensibilités/réactions à la perfusion était de 4,4% (123/2787), incluant 6 cas de Grade 3 et 3 cas de Grade 4.
Chez les patients traités par nivolumab 3 mg/kg en association à l'ipilimumab 1 mg/kg dans le MPM, l'incidence des hypersensibilités/réactions à la perfusion était de 12% (36/300). Des cas de Grade 2 et de Grade 3 ont été rapportés chez respectivement 5,0% (15/300) et 1,3% (4/300) des patients.
Anomalies des valeurs biologiques
Chez les patients traités par nivolumab en monothérapie, la proportion de patients ayant présenté une modification des paramètres biologiques par rapport à l'inclusion vers une anomalie de Grade 3 ou 4 a été la suivante : 5,4% pour les anémies (toutes de Grade 3), 0,9% pour les thrombocytopénies, 1,0% pour les leucopénies, 10,6% pour les lymphopénies, 1,1% pour les neutropénies, 2,3% pour les augmentations du taux de phosphatases alcalines, 3,0% pour les augmentations du taux d'ASAT, 2,5% pour les augmentations du taux d'ALAT, 1,3% pour les augmentations du taux de bilirubine totale, 0,8% pour les augmentations du taux de créatinine, 3,8% pour les hyperglycémies, 1,4% pour les hypoglycémies, 3,5% pour les augmentations de l'amylase, 7,9% pour les augmentations de la lipase, 6,7% pour les hyponatrémies, 1,7% pour les hyperkaliémies, 1,6% pour les hypokaliémies, 1,6% pour les hypercalcémies, 0,7% pour les hypermagnésémies, 0,5% pour les hypomagnésémies, 0,6% pour les hypocalcémies et 0,1% pour les hypernatrémies.
Chez les patients traités par nivolumab 3 mg/kg en association à l'ipilimumab 1 mg/kg dans le MPM, la proportion de patients ayant présenté une aggravation des paramètres biologiques par rapport à l'inclusion vers une anomalie de Grade 3 ou 4 a été la suivante : 2,4% pour les anémies, 1,0% chacune pour les thrombocytopénies et les leucopénies, 8,4% pour les lymphopénies, 1,3% pour les neutropénies, 3,1% pour les augmentations du taux de phosphatases alcalines, 7,1% chacune pour les augmentations du taux d'ASAT et du taux d'ALAT, 1,7% pour les augmentations du taux de bilirubine totale, 0,3% pour les augmentations du taux de créatinine, 2,8% pour les hyperglycémies, 5,4% pour les augmentations de l'amylase, 12,8% pour l'augmentation de la lipase, 0,7% pour les hypernatrémies, 8,1% pour les hyponatrémies, 4,1% pour les hyperkaliémies, 2,0% pour les hypokaliémies et 0,3% pour les hypocalcémies.
Immunogénicité
Parmi les 2234 patients traités par nivolumab en monothérapie à la dose de 3 mg/kg ou 240 mg toutes les 2 semaines, et évaluables pour la présence d'anticorps-anti-médicament, 255 patients (10,9%) étaient positifs au test de détection d'anticorps anti-médicaments avec quinze patients (0,6%) testés positifs pour des anticorps neutralisants.
Chez les patients traités par nivolumab en association à l'ipilimumab et évaluables pour la présence d'anticorps anti-nivolumab, l'incidence des anticorps anti-nivolumab était de 26,0% avec nivolumab 3 mg/kg et ipilimumab 1 mg/kg toutes les 3 semaines, 25,7% avec nivolumab 3mg/kg toutes les 2 semaines et ipilimumab 1 mg/kg toutes les 6 semaines, et de 37,8% avec nivolumab 1 mg/kg et ipilimumab 3 mg/kg toutes les 3 semaines. L'incidence des anticorps neutralisants dirigés contre nivolumab était de 0,5% avec nivolumab 3 mg/kg et ipilimumab 1 mg/kg toutes les 3 semaines, 0,7% avec nivolumab 3mg/kg toutes les 2 semaines et ipilimumab 1 mg/kg toutes les 6 semaines, et de 4,6% avec nivolumab 1 mg/kg et ipilimumab 3 mg/kg toutes les 3 semaines. Chez les patients dont il est possible d'évaluer la présence des anticorps anti-ipilimumab, l'incidence des anticorps anti- ipilimumab allait de 6,3 à 13,7%, et celle des anticorps neutralisants dirigés contre ipilimumab allait de 0 à 0,4%.
Bien que la clairance de nivolumab ait été augmentée de 20% lorsque les anticorps anti-nivolumab étaient présents, il n'a pas été mis en évidence de perte d'efficacité ou d'altération du profil de toxicité avec la présence d'anticorps nivolumab, basé sur les analyses de pharmacocinétique et de dose- réponse pour les deux traitements en monothérapie et en association.
Patients âgés
Aucune différence globale de tolérance n'a été rapportée entre les patients âgés (³ 65 ans) et les patients plus jeunes (< 65 ans).
Dans le MPM, il y a eu un taux plus important d'effets indésirables graves et d'arrêts de traitement dus à un effet indésirable chez les patients âgés de 75 ans ou plus (68% et 35% respectivement) comparé à tous les patients ayant reçu du nivolumab en association à l'ipilimumab (54% et 28% respectivement).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé doivent déclarer tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : signalement.social-sante.gouv.fr.
SURVEILLANCE du traitement : surveillance continue des effets indésirables cardiaques et pulmonaires, ainsi que pour des signes cliniques, des symptômes et des anomalies biologiques indiquant des troubles électrolytiques et une déshydratation, avant l'initiation du traitement et à intervalles réguliers au cours du traitement.
Remettre au patient la Carte d'Alerte Patient à chaque prescription et discuter avec le patient risques liés au traitement par ce médicament.
Grossesse
Aucune donnée sur l'utilisation de nivolumab n'est disponible chez la femme enceinte. Les études effectuées chez l'animal ont mis en évidence une toxicité embryofoetale (voir rubrique Données de sécurité précliniques). L'IgG4 humaine est connue pour traverser la barrière placentaire et nivolumab est une IgG4; par conséquent, il existe un risque potentiel de transmission de nivolumab de la mère vers le foetus. L'utilisation de nivolumab n'est pas recommandée chez la femme enceinte ou en âge de procréer n'utilisant pas de méthode efficace de contraception, à moins que le bénéfice clinique attendu ne dépasse le risque potentiel. Une méthode efficace de contraception doit être utilisée pendant toute la durée du traitement et poursuivie pendant 5 mois après la dernière perfusion de nivolumab.
Allaitement
On ne sait pas si nivolumab est excrété dans le lait maternel. Etant donné que de nombreux médicaments, y compris les anticorps, peuvent être excrétés dans le lait humain, un risque pour les nouveau-nés/nourrissons ne peut être exclu. Une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre le traitement par nivolumab en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la femme.
Fertilité
Les études permettant d'évaluer l'effet de nivolumab sur la fertilité n'ont pas été effectuées. En conséquence, l'effet de nivolumab sur la fertilité masculine et féminine n'est pas connu.
Nivolumab est un anticorps monoclonal humain, en conséquence aucune étude pharmacocinétique d'interaction n'a été réalisée. Les anticorps monoclonaux humains n'étant pas métabolisés par les enzymes du cytochrome P450 (CYP) ou d'autres enzymes métabolisant les médicaments, l'inhibition ou l'induction de ces enzymes lors de la co-administration de médicaments ne devraient pas entraîner de modification des paramètres pharmacocinétiques de nivolumab.
Autres formes d'interactions
Immunosuppression systémique
L'utilisation au préalable de corticoïdes systémiques et d'autres immunosuppresseurs doit être évitée avant de commencer nivolumab, du fait de leur interférence potentielle avec l'activité pharmacodynamique. Cependant, les corticoïdes systémiques et d'autres agents immunosuppresseurs peuvent être utilisés après l'initiation de nivolumab pour traiter les effets indésirables d'origine immunologique. Les résultats préliminaires montrent qu'une immunosuppression systémique après le début du traitement par nivolumab ne semble pas empêcher la réponse au nivolumab.
Le traitement doit être instauré et surveillé par un médecin expérimenté dans le traitement du cancer.
Posologie
OPDIVO en association à l'ipilimumab Mésothéliome Pleural Malin
La dose recommandée est de 360 mg de nivolumab administrée par voie intraveineuse pendant 30 minutes toutes les 3 semaines, en association avec 1 mg/kg d'ipilimumab administrée par voie intraveineuse pendant 30 minutes toutes les 6 semaines. Le traitement est poursuivi jusqu'à 24 mois chez les patients sans progression de la maladie.
Durée du traitement
Le traitement par OPDIVO, que ce soit en monothérapie ou en association à l'ipilimumab, doit être poursuivi tant qu'un bénéfice clinique est observé ou jusqu'à ce que le patient ne puisse plus tolérer le traitement (et jusqu'à la durée maximale du traitement si celle-ci est spécifiée pour une indication).
Des réponses atypiques (c'est à dire une augmentation initiale transitoire de la taille de la tumeur ou l'apparition de nouvelles petites lésions au cours des premiers mois, suivi de réduction de la tumeur) ont été observées. Il est recommandé de continuer le traitement par nivolumab en association à l'ipilimumab chez les patients cliniquement stables présentant des signes initiaux de progression de la maladie jusqu'à ce que la progression de la maladie soit confirmée.
Les augmentations ou diminutions de doses ne sont pas recommandées. Des administrations différées ou des interruptions de traitement peuvent être nécessaires selon la tolérance individuelle et la tolérabilité au traitement. Les recommandations concernant l'arrêt définitif du traitement ou la suspension des doses sont décrites dans le Tableau 1. Des recommandations détaillées de la prise en charge des effets indésirables d'origine immunologique sont décrites à la rubrique Mises en garde et précautions d'emploi.![]() Tableau 1 : Recommandations de modification du traitement par OPDIVO ou OPDIVO en association à l'ipilimumab
Tableau 1 : Recommandations de modification du traitement par OPDIVO ou OPDIVO en association à l'ipilimumab
Effet indésirable d'origine immunologique
Pneumopathie inflammatoire d'origine immunologique
Sévérité Modification de traitement
Pneumopathie de Grade 2 Suspendre la(les) dose(s) jusqu'à la résolution des symptômes, l'amélioration des anomalies radiographiques, et la fin du traitement par corticoïdes
Pneumopathie de Grade 3 ou
4
Arrêt définitif du traitement
Diarrhée ou colite de Grade 2 Suspendre la(les) dose(s) jusqu'à la résolution des symptômes et la fin du traitement par corticoïdes, s'il s'est avéré nécessaire
Colite d'origine immunologique
Diarrhée ou colite de Grade 3
- OPDIVO en monothérapie Suspendre la(les) dose(s)
jusqu'à la résolution des symptômes et la fin du traitement par corticoïdes
- OPDIVO+ipilimumab Arrêt définitif du traitement
Diarrhée ou colite de Grade 4 Arrêt définitif du traitement
Hépatite d'origine immunologique
Elévation de Grade 2 des aspartate aminotransférases (ASAT), des alanine aminotransférases (ALAT), ou de la bilirubine totale.
Suspendre la(les) dose(s) jusqu'au retour des valeurs biologiques aux valeurs initiales et jusqu'à la fin du traitement par corticoïdes, s'il s'est avéré nécessaire
Néphrite et
dysfonction rénale d'origine immunologique
Elévation de Grade 3 ou 4 des ASAT, ALAT, ou de la bilirubine totale
Elévation de la créatininémie de Grade 2 ou 3
Arrêt définitif du traitement
Suspendre la(les) dose(s) jusqu'au retour de la créatininémie à la valeur initiale et jusqu'à la fin du traitement par corticoïdes
![]() Elévation de la créatininémie de Grade 4 Arrêt définitif du traitement
Elévation de la créatininémie de Grade 4 Arrêt définitif du traitement
Endocrinopathies d'origine immunologique
Hypothyroïdie, hyperthyroïdie, hypophysite symptomatiques de Grade 2 ou 3,
Insuffisance surrénalienne de Grade 2 Diabète de Grade 3
Suspendre la(les) dose(s) jusqu'à la résolution des symptômes et la fin du traitement par corticoïdes (s'il s'est avéré nécessaire pour les symptômes d'une inflammation
![]() Tableau
1 : Recommandations de modification du
traitement par OPDIVO ou OPDIVO en association à l'ipilimumab
Tableau
1 : Recommandations de modification du
traitement par OPDIVO ou OPDIVO en association à l'ipilimumab
Hypothyroïdie de Grade 4 Hyperthyroïdie de Grade 4 Hypophysite de Grade 4
![]() Insuffisance surrénalienne de Grade 3 ou 4 Diabète de Grade 4
Insuffisance surrénalienne de Grade 3 ou 4 Diabète de Grade 4
aiguë). Le traitement doit être maintenu en cas de traitement substitutif hormonala tant qu'il n'y a pas de présence de symptômes
Arrêt définitif du traitement
Effets indésirables
cutanés d'origine immunologique
Eruption cutanée de Grade 3 Suspendre la(les) dose(s) jusqu'à la résolution des symptômes et la fin du traitement par corticoïdes
Eruption cutanée de Grade 4 Arrêt définitif du traitement
![]() Syndrome de Stevens-Johnson (SJS) ou nécrolyse épidermique toxique (NET)
Syndrome de Stevens-Johnson (SJS) ou nécrolyse épidermique toxique (NET)
Arrêt définitif du traitement (voir rubrique Mises en garde et précautions d'emploi)
Myocardite d'origine immunologique
Myocardite de Grade 2 Suspendre la(les) dose(s) jusqu'à la résolution des symptômes et la fin du traitement par corticoïdesb c
Myocardite de Grade 3 ou 4 Arrêt définitif du traitement
Grade 3 (première apparition) Suspendre la(les) dose(s)
Autres effets indésirables d'origine immunologique
Grade 4 ou Grade 3 récidivant ; Grade 2 ou 3 persistant malgré une modification de traitement ; impossibilité de réduire la dose de corticoïdes à 10 mg de prednisone ou équivalent par jour
Arrêt définitif du traitement
![]()
Note : Les grades de toxicité correspondent à la classification du National Cancer Institute Common Terminology Criteria for Adverse Events Version 4.0 (NCI-CTCAE v4).
a La recommandation pour l'utilisation d'un traitement substitutif hormonal est fournie en rubrique Mises en garde et précautions d'emploi.
b La tolérance de la reprise du traitement par nivolumab ou par nivolumab en association à l'ipilimumab, chez les patients ayant présenté précédemment une myocardite d'origine immunologique, n'est pas connue.
OPDIVO ou OPDIVO en association à l'ipilimumab doit être définitivement arrêté en cas de :
· Effets indésirables de Grade 4 ou de Grade 3 récidivants ;
· Effets indésirables de Grade2 ou 3 persistants malgré leur prise en charge.
Les patients traités par OPDIVO doivent recevoir la Carte d'Alerte Patient et être informés sur les risques liés à l'utilisation d'OPDIVO (voir également la notice).
Lorsqu'OPDIVO est administré en association à l'ipilimumab, si l'un des traitements est suspendu, l'autre traitement devra aussi être suspendu. Si l'administration est reprise après un temps différé, soit le traitement en association ou OPDIVO en monothérapie peut être repris sur la base de l'évaluation individuelle du patient.
Populations particulières
Population pédiatrique
La sécurité et l'efficacité d'OPDIVO chez les enfants de moins de 18 ans n'ont pas été établies. Aucune donnée n'est disponible.
Patients âgés
Aucune adaptation posologique n'est nécessaire chez les patients âgés (≥ 65 ans) (voir rubrique Propriétés pharmacocinétiques).
Insuffisance rénale
Sur la base des résultats de pharmacocinétique (PK) de population, aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance rénale légère ou modérée (voir rubrique Propriétés pharmacocinétiques). Les données chez les patients présentant une insuffisance rénale sévère sont trop limitées pour tirer des conclusions dans cette population.
Insuffisance hépatique
Sur la base des résultats de PK de population, aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère (voir rubrique Propriétés pharmacocinétiques). Les données chez les patients présentant une insuffisance hépatique modérée ou sévère sont trop limitées pour tirer des conclusions dans ces populations. OPDIVO doit être administré avec précaution chez les patients présentant une insuffisance hépatique modérée (bilirubine totale > 1,5 à 3 fois la limite supérieure de la normale [LSN], quel que soit le taux des ASAT) ou sévère (bilirubine totale > 3 fois LSN, quel que soit le taux des ASAT).
Mode d'administration
OPDIVO doit être exclusivement administré par voie intraveineuse. Il doit être administré en perfusion intraveineuse sur une période de 30 minutes. La perfusion doit être administrée à l'aide d'un filtre en ligne stérile, apyrogène, à faible liaison aux protéines (diamètre des pores de 0,2 µm à 1,2 µm).
OPDIVO ne doit pas être administré en intraveineux direct ni en bolus IV.
La dose totale d'OPDIVO requise peut être perfusée directement sans dilution à la concentration de 10 mg/mL en administration intraveineuse ou peut être diluée dans une solution injectable de chlorure de sodium à 9 mg/mL (0,9%) ou de glucose à 50 mg/mL (5%) (voir rubrique Instructions pour l'utilisation, la manipulation et l'élimination).
Lorsqu'il est administré en association à l'ipilimumab, OPDIVO doit être administré en premier suivi par l'ipilimumab le même jour. Utiliser des poches et des filtres de perfusion distincts pour chaque perfusion.
Pour les instructions concernant la préparation et la manipulation du médicament avant administration, voir la rubrique Instructions pour l'utilisation, la manipulation et l'élimination.
Durée de conservation :
Flacon non ouvert 3 ans.
Après ouverture
D'un point de vue microbiologique, une fois ouvert, le produit doit être perfusé ou dilué et perfusé immédiatement.
Après préparation de la perfusion
La stabilité chimique et physique en cours d'utilisation depuis le moment de la préparation a été démontrée comme suit (les délais incluent la période d'administration) :
| Préparation de la perfusion | Stabilité en cours d'utilisation | |
| Conservation entre 2ºC et 8ºC à l'abri de la lumière | Conservation à température ambiante (≤ 25°C) et à la lumière | |
| Non diluée ou diluée dans une solution injectable de chlorure de sodium à 9 mg/mL (0,9%) | 30 jours | 24 heures (sur un total de 30 jours de conservation) |
| Diluée dans une solution injectable de glucose à 50 mg/mL (5%) | 24 heures | 8 heures (sur un total de 24 heures de conservation) |
D'un point de vue microbiologique, la solution pour perfusion préparée, doit être utilisée immédiatement, indépendamment du diluant.
Si elle n'est pas utilisée immédiatement, les durées et les conditions de conservation après dilution et jusqu'à l'utilisation sont sous la responsabilité de l'utilisateur et ne doivent normalement pas dépasser 24 heures entre 2°C et 8°C ou 8 heures (sur un total de 24 heures de conservation) à température ambiante (≤ 25°C), à moins que la préparation de la perfusion n'ait été réalisée dans des conditions d'asepsie dûment contrôlées et validées.
Précautions particulières de conservation :A conserver au réfrigérateur (entre 2°C et 8°C). Ne pas congeler.
A conserver dans l'emballage extérieur d'origine, à l'abri de la lumière.
Le flacon non ouvert peut être conservé à température ambiante contrôlée jusqu'à 25°C et à la lumière ambiante jusqu'à 48 heures.
Pour les conditions de conservation après préparation de la perfusion, voir rubrique Durée de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments. OPDIVO ne doit pas être perfusé de manière concomitante avec d'autres médicaments, dans la même ligne de perfusion.
Aucun cas de surdosage n'a été rapporté dans les essais cliniques. En cas de surdosage, les patients doivent être étroitement surveillés à la recherche de signes ou symptômes évocateurs d'effets indésirables, et un traitement symptomatique approprié doit être instauré immédiatement.
Classe pharmacothérapeutique : Antinéoplasique, anticorps monoclonal. Code ATC : L01XC17.
Mécanisme d'action
Nivolumab est un anticorps monoclonal humain (HuMAb) de type immunoglobuline G4 (IgG4), qui se lie au récepteur PD-1 (programmed death-1) et bloque son interaction avec PD-L1 et PD-L2. Le récepteur PD-1 est un régulateur négatif de l'activité des cellules T, et il a été démontré qu'il est impliqué dans le contrôle de la réponse immunitaire des cellules T. La liaison du PD-1 avec les ligands PD-L1 et PD-L2, qui sont exprimés sur les cellules présentatrices d'antigène et peuvent être exprimés par les cellules tumorales ou par d'autres cellules du micro-environnement tumoral, entraîne une inhibition de la prolifération des cellules T et de la sécrétion de cytokines. Nivolumab potentialise les réponses des cellules T, incluant les réponses antitumorales, par un blocage de la liaison de PD-1 aux ligands PD-L1 et PD-L2. Dans des modèles syngéniques chez la souris, le blocage de l'activité du PD-1 a entraîné une diminution de la croissance de la tumeur.
L'inhibition médiée par nivolumab (anti-PD-1) associé à l'ipilimumab (anti-CTLA-4) entraîne l'amélioration des réponses anti-tumorales dans le mélanome métastatique. Dans des modèles de tumeurs murines syngéniques, le double blocage de PD-1 et CTLA-4 a entraîné une activité anti- tumorale synergique.
Efficacité et sécurité clinique
Sur la base d'un modèle de relations dose/réponse de l'efficacité et de la tolérance, il n'y a pas de différences cliniquement significatives en termes d'efficacité et de tolérance entre la posologie de nivolumab à 240 mg toutes les 2 semaines et à 3 mg/kg toutes les 2 semaines. De plus, sur la base de ces relations, il n'y a pas eu de différences cliniquement significatives entre la posologie de nivolumab à 480 mg toutes les 4 semaines et à 3 mg/kg toutes les 2 semaines dans le traitement adjuvant du mélanome, le mélanome avancé et le CCR (Carcinome à cellules rénales) avancé.
Mésothéliome pleural malin
Étude randomisée de phase 3 avec nivolumab en association à l'ipilimumab versus chimiothérapie (CA209743)
La tolérance et l'efficacité de nivolumab 3 mg/kg toutes les 2 semaines en association à l'ipilimumab 1 mg/kg toutes les 6 semaines ont été évaluées dans une étude randomisée de phase 3, en ouvert (CA209743). L'étude a inclus des patients (âgés de 18 ans ou plus) avec un mésothéliome pleural malin histologiquement confirmé et non préalablement traité, d'histologie épithélioïde ou non- épithélioïde, avec un statut de performance ECOG de 0 ou 1, et sans radiothérapie palliative dans les 14 jours précédant le premier traitement à l'étude. Les patients ont été inclus indépendamment du statut PD-L1 de leur tumeur.
Les patients présentant un mésothéliome primitif péritonéal, péricardique, ou de la tunique vaginale des testicules, une maladie pulmonaire interstitielle, une maladie auto-immune active, toute maladie nécessitant une immunosuppression systémique, et des métastases cérébrales (à moins d'une résection chirurgicale ou d'une radiothérapie stéréotaxique, et sans évolution dans les 3 mois précédant l'inclusion dans l'étude) ont été exclus de l'étude. La randomisation a été stratifiée en fonction de l'histologie (sous-types épithélioïdes vs. sarcomatoïdes ou histologie mixte) et du sexe (masculin vs. féminin).
Un total de 605 patients a été randomisé pour recevoir nivolumab en association à l'ipilimumab (n = 303) ou une chimiothérapie (n = 302). Les patients du bras nivolumab en association à l'ipilimumab ont reçu nivolumab 3 mg/kg, administré par voie intraveineuse pendant 30 minutes toutes les 2 semaines en association à l'ipilimumab 1 mg/kg, administré par voie intraveineuse pendant 30 minutes toutes les 6 semaines pour une durée maximale de 2 ans.
Les patients du bras chimiothérapie ont reçu une chimiothérapie pendant un maximum de 6 cycles (chaque cycle était de 21 jours). La chimiothérapie comprenait du cisplatine 75 mg/m² et du pemetrexed 500 mg/m², ou du carboplatine (ASC de 5) et du pemetrexed 500 mg/m².
Le traitement a été poursuivi jusqu'à progression de la maladie, toxicité inacceptable ou jusqu'à 24 mois. Le traitement pouvait se poursuivre après progression de la maladie si le patient était cliniquement stable et que l'investigateur considérait qu'il en tirait un bénéfice clinique. Les patients qui ont arrêté le traitement en association en raison d'un événement indésirable attribué à l'ipilimumab ont étaient autorisés à poursuivre le nivolumab en monothérapie. Les évaluations tumorales ont été réalisées toutes les 6 semaines après la première dose du traitement à l'étude pendant les 12 premiers mois, puis toutes les 12 semaines et ce jusqu'à progression de la maladie ou arrêt du traitement à l'étude.
Les caractéristiques des patients à l'inclusion dans l'étude CA209743 étaient généralement équilibrées entre les différents bras de traitement. L'âge médian était de 69 ans (de 25 à 89 ans), avec 72% ≥ 65 ans et 26% ≥ 75 ans. La majorité des patients était de type caucasien (85%) et de sexe masculin (77%). Le statut de performance ECOG à l'inclusion était de 0 (40%) ou de 1 (60%), 80% des patients présentaient un statut PD-L1 ≥ 1% et 20% un statut PD-L1 < 1%, 75% présentaient une histologie épithélioïde et 25% non épithélioïde.
Le critère d'évaluation principal d'efficacité de l'étude CA209743 était la SG. Les autres critères d'efficacité étaient la SSP, l'ORR, et la durée de réponse, évalués par un comité de revue indépendant centralisé en aveugle (Blinded Independent Central Review BICR), en utilisant les critères RECIST modifiés.
L'étude a démontré une amélioration statistiquement significative en SG pour les patients randomisés dans le bras nivolumab en association à l'ipilimumab comparé à la chimiothérapie, lors de l'analyse intermédiaire pré-spécifiée, après l'observation d'au moins 419 événements (89% du nombre prévu d'événements pour l'analyse finale). Le suivi minimum pour la SG était de 22 mois.
Les résultats d'efficacité sont présentés dans la figure 1 et dans le Tableau 5.
Figure 1: Courbes de Kaplan-Meier pour la SG CA209743)
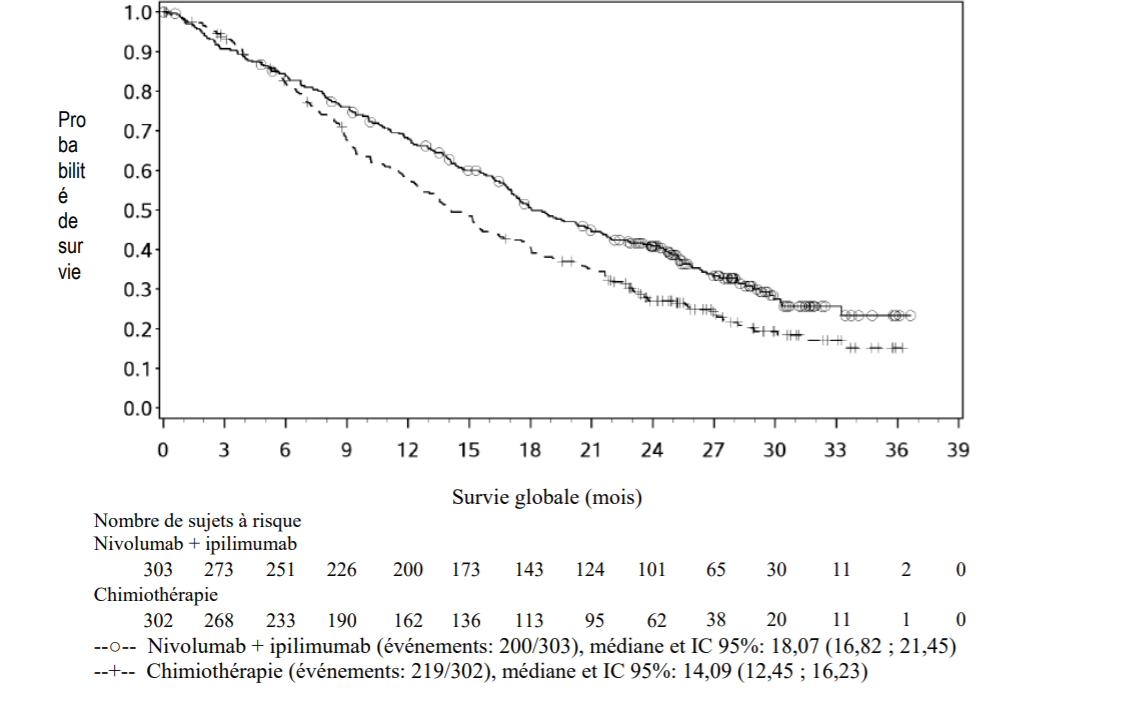
Tableau 5: Résultats d'efficacité (CA209743)
| | nivolumab + ipilimumab (n = 303) | chimiothérapie (n = 302) | ||
| Survie Globale Evénements | 200 (66%) | 219 (73%) | ||
| Hazard ratio | 0,74 | |||
| (96.6% CI)b | (0,60 ; 0,91) | |||
| Log-rank stratifié p-valuec | 0,002 | |||
| Médiane (mois)a | 18,1 | 14,1 | ||
| (IC 95%) | (16,8 ; 21,5) | (12,5 ; 16,2) | ||
| Taux (95% CI) à 24 moisa | 41% (35,1 ; 46,5) | 27% (21,9 ; 32,4) | ||
| Survie sans progression Evénements | 218 (72%) | 209 (69%) | ||
| Hazard ratio b | 1,0 | |||
| (IC 95%) | (0,82 ; 1,21) | |||
| Log-rank stratifié p-value | c | 0,0001 | ||
| Médiane (mois) (IC 95%) | a | | 6,8 | 7,2 |
| (5,6 ; 7,4) | (6,9 ; 8,1) | |||
| Taux de réponse objective | 40% | 43% | ||
| (IC 95%) | (34,1 ; 45,4) | (37,1 ; 48,5) | ||
| Réponse complète (RC) | 1,7% | 0 | ||
| Réponse partielle (RP) | 38% | 43% | ||
| Durée de réponse Médiane (mois)a (IC 95%) | 11,0 | 6,7 | ||
| (8,1 ; 16,5) | (5,3 ; 7,1) | |||
| % avec durée ≥ 6 mois | 69% | 53% | ||
a Estimation de Kaplan-Meier.
b Modèle à risques proportionnels de Cox stratifié.
c La valeur p est comparée à l'alpha attribué de 0,0345 pour cette analyse intermédiaire
44,2% et 40,7% des patients ont reçu un traitement systémique ultérieur dans les bras en association et chimiothérapie, respectivement. Une immunothérapie ultérieure (incluant anti-PD-1, anti-PD-L1, et anti-CTLA-4) a été reçue par 3,3% et 20,2% des patients dans les bras en association et chimiothérapie, respectivement.
Les résultats de SG chez les patients avec une expression tumorale de PD-L1 < 1% (HR [IC 95%] étaient de 0,94 [0,62 ; 1,40], n = 57), et de 0,69 [0,55 ; 0,87], n = 232) chez les patients avec une expression tumorale de PD-L1 ≥1% (HR [IC 95%].
Le tableau 6 résume les résultats d'efficacité pour la SG, la SSP et l'ORR par histologie dans les analyses pré-spécifiées en sous-groupes.
Tableau 6 : Résultats d'efficacité par histologie (CA209743)
| Epithélioïde (n = 471) | Non-épithélioïde (n = 134) | |||
| | nivolumab | chimiothérapie | nivolumab | chimiothérapie |
| | + | (n = 235) | + | (n = 67) |
| | ipilimumab | | ipilimumab | |
| | (n = 236) | | (n = 67) | |
| Survie globale | | | | |
| Evénements | 157 | 164 | 43 | 55 |
| Hazard ratio (95% CI)a | 0,85 (0,68; 1,06) | 0.46 (0,31 ; 0,70) | ||
| Médiane (mois)a | 18,73 | 16,23 | 16,89 | 8,80 |
| (IC 95%) | (17,05 ; 21,72) | (14,09 ; 19,15) | (11,83 ; 25,20) | (7,62 ; 11,76) |
| Taux (IC 95%) à 24 moisa | 41,2 (34,7 ; 47,6) | 31,8 (25,7 ; 38,1) | 39,5 (27,5 ; 51,2) | 9,7 (3,8; 18,9) |
| Survie sans progression | ||||
| Hazard ratio (IC 95%)a | 1,14 (0,92; 1,41) | 0,58 (0,38 ; 0,90) | ||
| Taux de réponse objective | 38,6% | 47,2% | 43,3% | 26,9% |
| (IC 95%) b | (32,3 ; 45,1) | (40,7 ; 53,8) | (31,2 ; 56,0) | (16,8 ; 39,1) |
| Durée de réponse | 8,44 | 6,83 | 24,02 | 4,21 |
| Médiane (mois) | (7,16 ; 14,59) | (5,59 ; 7,13) | (8,31 ; N.A.) | (2,79 ; 7,03) |
| (IC 95%)c | ||||
a Rapport des risques basé sur le modèle à risques proportionnels de Cox non stratifié
b Intervalle de confiance basé sur la méthode de Clopper et Pearson
c Médiane calculée selon la méthode de Kaplan-Meier
Un total de 157 patients atteints de MPM et âgés de ≥ 75 ans ont été inclus dans l'étude CA209743 (78 dans le bras nivolumab en association à l'ipilimumab et 79 dans le bras chimiothérapie). Un HR de 1,02 (IC 95% : 0,70 ; 1,48) en SG a été observé pour nivolumab en association à l'ipilimumab vs. chimiothérapie au sein de ce sous-groupe d'étude. Un taux plus élevé d'effets indésirables graves et d'arrêt de traitement dû à des effets indésirables a été montré chez les patients âgés de 75 ans ou plus comparé à l'ensemble des patients ayant reçu du nivolumab en association à l'ipilimumab (voir rubrique Effets indésirables). Cependant, compte tenu de la nature exploratoire de cette analyse en sous-groupe, aucune conclusion définitive ne peut être tirée.
Tolérance et efficacité chez les patients âgés
Aucune différence globale de tolérance ou d'efficacité n'a été rapportée entre les patients âgés (³ 65 ans) et les patients plus jeunes (< 65ans). Les données provenant des patients atteints de MPM ont montré un taux plus élevé d'effets indésirables graves et d'arrêt dû à des effets indésirables chez les patients de 75 ans ou plus (68% et 35% respectivement) comparé à tous les patients ayant reçu du nivolumab en associationà l'ipilimumab (54% et 28% respectivement).
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec le nivolumab dans un ou plusieurs sous-groupes de la population pédiatrique dans le traitement des tumeurs malignes solides, des tumeurs malignes du tissu lymphoïde et des tumeurs malignes du système nerveux central (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Nivolumab en monothérapie
La pharmacocinétique de nivolumab est linéaire pour des posologies allant de 0,1 à 10 mg/kg. La moyenne géométrique de la clairance (CL), la demi-vie terminale, et l'exposition moyenne à l'état d'équilibre à la dose de 3 mg/kg de nivolumab toutes les 2 semaines étaient respectivement de 7,9 mL/h, 25,0 jours, et 86,6 µg/mL, sur la base d'analyses de pharmacocinétique de population.
La voie métabolique de nivolumab n'a pas été caractérisée. Il est attendu que nivolumab soit dégradé en petits peptides et acides aminés via des voies cataboliques au même titre que les IgG endogènes.
Nivolumab en association à l'ipilimumab
Lorsque nivolumab 3 mg/kg était administré en association à ipilimumab 1 mg/kg, la clairance de nivolumab a augmenté de 1%, et la clairance de l'ipilimumab a été diminuée de 1,5%, ces changements n'étant pas considérés comme cliniquement pertinents.
Lorsqu'il est administré en association avec l'ipilimumab, la clairance de nivolumab est augmentée de 20% en présence d'anticorps anti-nivolumab et la clairance de l'ipilimumab est augmentée de 5,7% en présence d'anticorps anti-ipilimumab. Ces changements n'ont pas été considérés comme étant cliniquement pertinents.
Populations particulières
Une analyse pharmacocinétique de population suggère que les facteurs suivants n'avaient pas d'incidence sur la CL de nivolumab : âge, sexe, race, type de tumeur solide, taille de la tumeur et insuffisance hépatique. Bien que le statut ECOG, le taux de filtration glomérulaire (TFG) initial, le taux d'albumine, le poids corporel, et une insuffisance hépatique légère aient eu un effet sur la clairance de nivolumab, cet effet n'a pas été cliniquement significatif.
Insuffisance rénale
L'effet d'une insuffisance rénale sur la CL de nivolumab a été évalué chez des patients présentant une insuffisance rénale légère (TFG < 90 et ≥ 60 mL/min/1,73 m2; n = 379), modérée (TFG < 60 et ≥ 30 mL/min/1,73 m2; = 179), ou sévère (TFG <30 et ≥ 15 mL/min/1,73m2; n = 2) comparativement à des patients présentant une fonction rénale normale (TFG ≥ 90 mL/min/1,73 m2; n = 342) dans des analyses de pharmacocinétique de population. Il n'y a pas eu de différence de CL de nivolumab cliniquement importante entre les patients présentant une insuffisance rénale légère à modérée et les patients présentant une fonction rénale normale. Les données chez les patients avec une insuffisance rénale sévère sont trop limitées pour tirer des conclusions dans cette population (voir rubrique Posologie et mode d'administration).
Insuffisance hépatique
L'effet d'une insuffisance hépatique sur la CL de nivolumab a été évalué chez des patients présentant une insuffisance hépatique légère (bilirubine totale (BT) 1,0 × à 1,5 × LSN ou ASAT > LSN selon la définition des critères de l'insuffisance hépatique du NCI (National Cancer Institute); n = 92) comparativement aux patients présentant une fonction hépatique normale (BT et ASAT ≤ LSN; n = 804) dans des analyses de pharmacocinétique de population. Il n'y a pas eu de différence cliniquement importante de la CL de nivolumab entre les patients présentant une insuffisance hépatique légère et les patients présentant une fonction hépatique normale. Nivolumab n'a pas été étudié chez des patients présentant une insuffisance hépatique modérée (BT > 1,5 × à 3 × LSN et quel que soit le taux d'ASAT) ou sévère (BT > 3 × LSN et quel que soit le taux d'ASAT) (voir rubrique Posologie et mode d'administration).
Nivolumab ou nivolumab en association avec l'ipilimumab peut avoir une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines. Du fait des effets indésirables potentiels, tels que la fatigue, (voir rubrique Effets indésirables), les patients doivent être prévenus qu'ils doivent être prudents lorsqu'ils conduisent ou utilisent des machines, tant qu'ils ne sont pas certains que nivolumab n'altère pas leur vigilance.
Le blocage du signal PD-L1 a montré chez des modèles murins de grossesse une perturbation de la tolérance au foetus et une augmentation de la perte foetale. Les effets du nivolumab sur le développement pré et postnatal ont été évalués chez des singes recevant nivolumab 2 fois par semaine, du début de l'organogénèse, durant le premier trimestre, jusqu'à l'accouchement, à des niveaux d'exposition de 8 à 35 fois supérieurs à ceux associés à la dose clinique de nivolumab de 3 mg/kg (basés sur l'AUC). Il y a eu une augmentation dose-dépendante du nombre d'avortements et de mortalité néonatale à partir du troisième trimestre.
Le reste de la descendance des femelles traitées par nivolumab a survécu jusqu'au terme prévu, sans signes cliniques liés au traitement, ni anomalies du développement, ni altération du poids des organes ou modifications pathologiques macro ou microscopiques. Les résultats des courbes de croissance, ainsi que les données de tératogénicité, neurocomportementales, immunologiques et les paramètres de pathologie clinique analysés pendant une période post-natale de 6 mois ont été comparables avec ceux du groupe contrôle. Cependant, basé sur son mécanisme d'action, l'exposition foetale à nivolumab peut augmenter le risque de développer des troubles d'origine immunologique ou altérer la réponse immunitaire normale, et des troubles d'origine immunologique ont été rapportés chez des souris déficientes en PD-1.
Aucune étude de fertilité n'a été réalisée avec le nivolumab.
La préparation doit être réalisée par du personnel formé conformément aux règles de bonnes pratiques, particulièrement en ce qui concerne le respect de l'asepsie.
Préparation et administration
Calcul de la dose
Plus d'un flacon de solution à diluer d'OPDIVO peut être nécessaire pour administrer la dose totale au patient.
Nivolumab en association à l'ipilimumab dans le MPM
La dose prescrite pour le patient est de 360 mg, administrés indépendamment du poids du patient.
Préparation de la perfusion
S'assurer d'opérer dans des conditions aseptiques lorsque vous préparez la perfusion.
OPDIVO peut être utilisé en administration intraveineuse, soit :
- sans dilution, après transfert dans un récipient pour perfusion en utilisant une seringue stérile appropriée; ou
- après dilution, selon les instructions suivantes :
§ le volume total de la perfusion ne doit pas dépasser 160 mL. Pour les patients pesant moins de 40 kg, le volume total de la perfusion ne doit pas dépasser 4 mL par kilogramme de poids du patient.
La solution à diluer d'OPDIVO peut être diluée avec soit :
§ une solution injectable de chlorure de sodium à 9 mg/mL (0,9%) ; ou
§ une solution injectable de glucose à 50 mg/mL (5%)
ETAPE 1
- Inspecter la solution à diluer d'OPDIVO pour mettre en évidence la présence de particules étrangères ou d'une coloration anormale. Ne pas secouer le flacon. La solution à diluer d'OPDIVO est un liquide clair à opalescent, incolore à jaune pâle. Jeter le flacon si la solution est trouble, d'une coloration anormale, ou contient un type de particules autre que quelques particules translucides à blanches.
- Retirer le volume nécessaire de solution à diluer d'OPDIVO en utilisant une seringue stérile de volume approprié.
ETAPE 2
- Transférer la solution à diluer dans une bouteille en verre stérile et évacuée, ou dans un récipient pour perfusion (PVC ou polyoléfine).
- Le cas échéant, diluer avec le volume nécessaire de solution injectable de chlorure de sodium à 9 mg/mL (0,9%) ou de solution injectable de glucose à 50 mg/mL (5%). Afin de faciliter la préparation, la solution à diluer peut directement être transvasée dans une poche pré-remplie contenant le volume approprié de solution injectable de chlorure de sodium à 9 mg/mL (0,9%) ou de solution injectable de glucose à 50 mg/mL (5%).
- Mélanger doucement la perfusion par rotation manuelle. Ne pas secouer.
Administration
La perfusion d'OPDIVO ne doit pas être administrée en IV rapide ni en bolus IV. Administrer la perfusion d'OPDIVO par voie intraveineuse sur une période de 30 minutes.
La solution d'OPDIVO ne doit pas être perfusée simultanément avec d'autres médicaments sur la même ligne intraveineuse. Utiliser une ligne intraveineuse séparée pour la perfusion.
Utiliser un set de perfusion et un filtre en ligne stérile, apyrogène, à faible liaison aux protéines (diamètre des pores de 0,2 µm à 1,2 µm).
La perfusion d'OPDIVO est compatible avec les poches en PVC et en polyoléfine, les flacons en verre, les sets de perfusion en PVC, et les filtres en ligne avec membranes en polyethersulfone et tailles de pores de 0,2 µm à 1,2 µm.
Après administration de la dose de nivolumab, rincer la ligne de perfusion avec une solution injectable de chlorure de sodium à 9 mg/mL (0,9%) ou une solution injectable de glucose à 50 mg/mL (5%).
Elimination
Ne pas conserver toute fraction inutilisée de la solution pour perfusion en vue d'une réutilisation. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Médicament réservé à l'usage hospitalier.
Prescription réservée aux spécialistes en oncologie, ou aux médecins
compétents en cancérologie. Médicament nécessitant une surveillance
particulière pendant le traitement.
Solution à diluer pour perfusion (solution à diluer stérile).
Solution claire à opalescente, incolore à jaune pâle, pouvant contenir quelques particules légères. Le pH de la solution est approximativement de 6,0 et l'osmolarité approximativement de 340 mOsm/kg.
4 mL de solution à diluer stérile en flacon de 10 mL (verre de type I) muni d'un bouchon (caoutchouc butyl enrobé) et d'un opercule amovible (aluminium). Boîte de 1 flacon.
Chaque mL de solution à diluer pour perfusion contient 10 mg de nivolumab.
Un flacon de 4 mL contient 40 mg de nivolumab.
Nivolumab est produit sur des cellules ovariennes de hamster chinois, par la technologie de l'ADN recombinant.
Excipient à effet notoire :
Chaque mL de solution à diluer contient 0,1 mmol (ou 2,5 mg) de sodium. Pour la liste complète des excipients, voir rubrique Liste des excipients.
Citrate de sodium dihydraté
Chlorure de sodium
Mannitol (E421)
Acide pentétique (acide diéthylène-triamine-penta-acétique)
Polysorbate 80 (E433)
Hydroxyde de sodium (pour ajustement du pH)
Acide chlorhydrique (pour ajustement du pH)
Eau pour préparations injectables