
SAXENDA 6 mg-ml, solution injectable en stylo prérempli, boîte de 3 stylos préremplis de 3 ml
Dernière révision : 22/11/2024
Taux de TVA : 10%
Laboratoire exploitant : NOVO NORDISK
Source :

Adultes
Saxenda est
indiqué en complément d'un régime hypocalorique et d'une augmentation de
l'activité physique dans le contrôle du poids chez des patients adultes ayant
un Indice de Masse Corporelle (IMC) initial :
● ≥
30 kg/m² (obésité), ou
● ≥
27 kg/m² et < 30 kg/m² (surpoids) en présence d'au moins un facteur de
comorbidité lié au poids tel qu'une dysglycémie (prédiabète ou diabète de type
2), une hypertension artérielle, une dyslipidémie ou un syndrome d'apnée
obstructive du sommeil.
Le traitement par Saxenda doit être interrompu après 12 semaines à la dose de 3,0 mg/jour si les patients n'ont pas perdu au moins 5 % de leur poids initial.
Adolescents (≥ 12 ans)
Saxenda peut
être utilisé en complément d'une alimentation saine et d'une augmentation de
l'activité physique dans le contrôle du poids chez des patients adolescents à
partir de 12 ans ayant :
● une
obésité (correspondant à un IMC ≥ 30 kg/m² chez les adultes selon les
seuils internationaux)* et
● un
poids corporel supérieur à 60 kg.
Le traitement par Saxenda doit être interrompu et réévalué si les patients n'ont pas perdu au moins 4 % de leur IMC ou de leur Z-score d'IMC après 12 semaines à la dose de 3,0 mg/jour ou à la dose maximale tolérée.
* Seuils d'IMC selon l'IOTF pour l'obésité en fonction du sexe entre 12 et 18 ans (voir tableau 1) :
Tableau 1
Seuils d'IMC selon l'IOTF pour l'obésité en fonction du sexe entre 12 et 18 ans
|
Âge
(années) |
IMC correspondant à 30 kg/m² pour les adultes selon les seuils internationaux. | |
| Garçons | Filles | |
| 12 | 26,02 | 26,67 |
| 12,5 | 26,43 | 27,24 |
| 13 | 26,84 | 27,76 |
| 13,5 | 27,25 | 28,20 |
| 14 | 27,63 | 28,57 |
| 14,5 | 27,98 | 28,87 |
| 15 | 28,30 | 29,11 |
| 15,5 | 28,60 | 29,29 |
| 16 | 28,88 | 29,43 |
| 16,5 | 29,14 | 29,56 |
| 17 | 29,41 | 29,69 |
| 17,5 | 29,70 | 29,84 |
| 18 | 30,00 | 30,00 |
Hypersensibilité au liraglutide ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Aspiration pulmonaire en association avec une anesthésie générale ou une sédation profonde
Des cas d'aspiration pulmonaire du contenu gastrique ont été signalés chez des patients recevant des agonistes des récepteurs du GLP-1 subissant une anesthésie générale ou une sédation profonde. Par conséquent, le risque accru de contenu gastrique résiduel en raison du retard de vidange gastrique (voir rubrique Effets indésirables) doit être pris en considération avant de réaliser des procédures impliquant une anesthésie générale ou une sédation profonde.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Patients présentant une insuffisance cardiaque
Il n'y a pas d'expérience clinique chez les patients présentant une insuffisance cardiaque congestive de classe IV New York Heart Association (NYHA). L'utilisation du liraglutide n'est donc pas recommandée chez ces patients.
Populations particulières
La sécurité et l'efficacité du liraglutide dans le contrôle du poids n'ont pas été établies chez les patients :· âgés
de 75 ans ou plus,
· traités par d'autres produits utilisés pour contrôler le poids,
· présentant une obésité secondaire à des troubles endocrinologiques, à des troubles alimentaires ou à des traitements médicamenteux susceptibles d'entraîner une prise de poids,
· présentant une insuffisance rénale sévère,
· présentant une insuffisance hépatique sévère.
L'utilisation
chez ces patients n'est pas recommandée (voir rubrique Posologie et mode
d'administration).
· traités par d'autres produits utilisés pour contrôler le poids,
· présentant une obésité secondaire à des troubles endocrinologiques, à des troubles alimentaires ou à des traitements médicamenteux susceptibles d'entraîner une prise de poids,
· présentant une insuffisance rénale sévère,
· présentant une insuffisance hépatique sévère.
Comme le liraglutide n'a pas fait l'objet d'investigations dans le contrôle du poids chez les sujets présentant une insuffisance hépatique légère ou modérée, il doit être utilisé avec précaution chez ces patients (voir rubriques Posologie et mode d'administration et Propriétés pharmacocinétiques).
L'expérience chez les patients présentant une maladie inflammatoire de l'intestin et une gastroparésie diabétique est limitée. L'utilisation du liraglutide n'est pas recommandée chez ces patients car elle est associée à des réactions indésirables gastro-intestinales passagères telles que nausées, vomissements et diarrhées.
Pancréatite
Des cas de pancréatites aigues ont été observés lors de l'utilisation d'agonistes des récepteurs du GLP-1. Les patients doivent être informés des symptômes caractéristiques de la pancréatite aiguë. En cas de suspicion de pancréatite, le liraglutide doit être arrêté ; si une pancréatite aiguë est confirmée, le liraglutide ne doit pas être réadministré.
Cholélithiase et cholécystite
Lors des essais cliniques dans le contrôle du poids, une fréquence plus élevée des cas de cholélithiase et de cholécystite a été observée chez les patients traités par le liraglutide que chez les patients sous placebo. Le fait qu'une perte de poids importante puisse augmenter le risque de cholélithiase et donc de cholécystite n'expliquait que partiellement leur fréquence accrue dans le groupe traité par le liraglutide. La cholélithiase et la cholécystite peuvent conduire à une hospitalisation et une cholécystectomie. Les patients doivent être informés des symptômes caractéristiques de la cholélithiase et de la cholécystite.
Maladie thyroïdienne
Lors des essais cliniques sur le diabète de type 2, des évènements indésirables thyroïdiens, tel qu'un goitre, ont été rapportés en particulier chez les patients présentant une maladie thyroïdienne préexistante. Le liraglutide doit donc être utilisé avec précaution chez les patients présentant une maladie thyroïdienne.
Fréquence cardiaque
Une augmentation de la fréquence cardiaque a été observée avec le liraglutide lors des essais cliniques (voir rubrique Propriétés pharmacodynamiques). La fréquence cardiaque doit être contrôlée à intervalles réguliers conformément à la pratique clinique habituelle. Les patients doivent être informés des symptômes associés à une augmentation du rythme cardiaque (palpitations ou sensations d'emballement du cœur à l'état de repos). Le traitement par le liraglutide doit être arrêté chez les patients qui présentent une élévation durable cliniquement significative de la fréquence cardiaque au repos.
Déshydratation
Des signes et des symptômes de déshydratation, incluant une insuffisance rénale et une insuffisance rénale aiguë, ont été rapportés chez des patients traités par des agonistes des récepteurs du GLP-1. Les patients traités par liraglutide doivent être avertis du risque potentiel de déshydratation liée aux effets indésirables gastro-intestinaux et doivent prendre des précautions pour éviter une perte hydrique.
Hypoglycémie chez les patients ayant un diabète de type 2
Les patients ayant un diabète de type 2 traités par le liraglutide en association à une insuline et/ou à un sulfamide hypoglycémiant peuvent présenter une augmentation du risque d'hypoglycémie. Le risque d'hypoglycémie peut être diminué en réduisant la dose d'insuline et/ou du sulfamide hypoglycémiant.
Population pédiatrique
Des épisodes d'hypoglycémie cliniquement significative ont été rapportés chez des adolescents (≥ 12 ans) traités par le liraglutide. Les patients doivent être informés des symptômes caractéristiques de l'hypoglycémie et de la conduite à tenir.
Hyperglycémie chez les patients ayant un diabète traité à l'insuline
Chez les patients diabétiques, Saxenda ne doit pas être utilisé comme un substitut à l'insuline. Une acidocétose diabétique a été rapportée chez des patients insulino-dépendants après un arrêt brutal du traitement ou une réduction de la dose d'insuline (voir rubrique Posologie et mode d'administration).
Excipients
Saxenda contient moins de 1 mmol de sodium (23 mg) par dose ; le médicament est donc essentiellement « sans sodium ».
Résumé du profil de sécurité:
La sécurité de Saxenda a été évaluée au cours de 5 essais en double aveugle, contrôlés versus placebo dans lesquels ont été inclus 5 813 patients adultes avec un surpoids ou une obésité présentant au moins une comorbidité liée au poids. Dans l'ensemble, les réactions indésirables le plus fréquemment rapportées lors du traitement par Saxenda étaient les réactions gastro-intestinales (67,9 %) (voir rubrique « Description de certaines réactions indésirables »).
Liste tabulée des réactions indésirables
Le tableau 3 répertorie les réactions indésirables observées chez les adultes. Les réactions indésirables sont listées par classe de systèmes d'organes et par fréquence. Les catégories de fréquences sont définies de la manière suivante : très fréquent (≥ 1/10); fréquent (≥ 1/100, < 1/10); peu fréquent (≥ 1/1 000, < 1/100); rare (≥ 1/10 000, < 1/1 000); très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les réactions indésirables sont présentées suivant un ordre décroissant de gravité.
Tableau 3 Réactions indésirables rapportées chez les adultes
|
Classes
de systèmes d'organes MedDRA |
Très fréquent | Fréquent | Peu fréquent | Rare |
Fréquence
indéterminée |
|
Affections
du système immunitaire |
Réaction anaphylactique | ||||
|
Troubles
du métabolisme et de la nutrition |
Hypoglycémie* | Déshydratation | |||
| Affections psychiatriques | Insomnie** | ||||
|
Affections
du système nerveux |
Maux de tête |
Vertiges
Dysgueusie |
|||
| Affections cardiaques | Tachycardie | ||||
|
Affections
gastro- intestinales |
Nausées
Vomissements Diarrhées Constipation |
Sécheresse
buccale Dyspepsie Gastrite Reflux gastro-œsophagien Douleur abdominale haute Flatulences Éructation Distension abdominale |
Pancréatite***
Vidange gastrique retardée**** |
Obstruction intestinale† | |
|
Affections
hépatobiliaires |
Lithiase biliaire*** | Cholécystite*** | |||
|
Affections
de la peau et du tissu sous-cutané |
Eruption cutanée | Urticaire | Amyloïdose cutanée |
||
|
Affections
du rein et des voies urinaires |
Insuffisance
rénale aiguë Insuffisance rénale |
||||
|
Troubles
généraux et anomalies au site d'administration |
Réactions
au site d'injection Asthénie Fatigue |
Malaise | |||
| Investigations |
Lipase
augmentée Amylase augmentée |
*Hypoglycémie (basée sur les
symptômes rapportés par les patients eux-mêmes et non confirmée par une mesure
de la glycémie) rapportée chez les patients ne présentant pas de diabète de
type 2 traités par Saxenda en association à un régime alimentaire et de
l'activité physique. Voir rubrique « Description de certaines réactions
indésirables » pour plus d'informations.
**L'insomnie a été principalement observée pendant les 3 premiers mois de traitement.
***Voir rubrique Mises en garde spéciales et précautions d'emploi.
****issu d'essais cliniques contrôlés de phase 2, 3a et 3b.
†Effets indésirables issus des sources post-commercialisation
**L'insomnie a été principalement observée pendant les 3 premiers mois de traitement.
***Voir rubrique Mises en garde spéciales et précautions d'emploi.
****issu d'essais cliniques contrôlés de phase 2, 3a et 3b.
†Effets indésirables issus des sources post-commercialisation
Description de certaines réactions indésirables:
Hypoglycémie
chez les patients ne présentant pas de diabète de type 2
Lors des
essais cliniques menés chez des patients obèses ou en surpoids sans diabète de
type 2 et traités par Saxenda en association à un régime alimentaire et de
l'activité physique, aucun événement hypoglycémique sévère (nécessitant
l'intervention d'un tiers) n'a été rapporté. Des symptômes d'hypoglycémie ont
été rapportés par 1,6 % des patients traités par Saxenda et par 1,1 % des
patients du groupe placebo ; néanmoins, ces événements n'ont pas été confirmés
par une mesure de la glycémie. La majorité de ces événements était d'intensité
légère.
Hypoglycémie
chez les patients ayant un diabète de type 2
Lors d'un
essai clinique mené chez des patients obèses ou en surpoids ayant un diabète de
type 2 et traités par Saxenda en association à un régime alimentaire et de
l'activité physique, une hypoglycémie sévère (nécessitant l'intervention d'un
tiers) a été rapportée par 0,7 % des patients traités par Saxenda et uniquement
chez les patients traités de façon concomitante par sulfamide hypoglycémiant.
De plus, chez ces patients, une hypoglycémie symptomatique documentée a été
rapportée par 43,6 % des patients traités par Saxenda et par 27,3 % des
patients recevant un placebo. Parmi les patients qui ne prenaient pas de
traitement concomitant par sulfamide hypoglycémiant, 15,7 % des patients
traités par Saxenda et 7,6 % des patients recevant un placebo ont rapporté des
événements hypoglycémiques symptomatiques documentés (définis par une glycémie ≤
3,9 mmol/l et la présence de symptômes).
Hypoglycémie chez les patients ayant un diabète de type 2
traités par une insuline
Lors
d'un essai clinique mené chez des patients obèses ou en surpoids ayant un
diabète de type 2 et traités par une insuline et liraglutide 3,0 mg/jour en
association à un régime alimentaire, de l'activité physique et jusqu'à 2
antidiabétiques oraux, une hypoglycémie sévère (nécessitant l'intervention d'un
tiers) a été rapportée par 1,5 % des patients traités par liraglutide 3,0
mg/jour. Dans cette étude, une hypoglycémie symptomatique documentée (définie
par une glycémie ≤ 3,9 mmol/l et la présence de symptômes) a été
rapportée par 47,2 % des patients traités par liraglutide 3,0 mg/jour et par
51,8 % des patients recevant un placebo. Parmi les patients traités de façon
concomitante par sulfamide hypoglycémiant, 60,9 % des patients traités par
liraglutide 3,0 mg/jour et 60,0 % des patients recevant un placebo ont rapporté
des événements hypoglycémiques symptomatiques documentés.
Réactions
indésirables gastro-intestinales
La majorité
des événements gastro-intestinaux était d'intensité légère à modérée,
transitoire et n'a pas nécessité l'arrêt du traitement. Ces réactions
survenaient généralement pendant les premières semaines de traitement et
diminuaient au bout de quelques jours ou semaines de poursuite du traitement.
Les patients ≥ 65 ans peuvent être davantage sujets aux effets indésirables gastro-intestinaux lorsqu'ils sont traités par Saxenda.
Les patients présentant une insuffisance rénale légère ou modérée (clairance de la créatinine ≥ 30 ml/min) peuvent être davantage sujets aux effets gastro-intestinaux lorsqu'ils sont traités par Saxenda.
Insuffisance
rénale aiguë
Des cas
d'insuffisance rénale aiguë ont été rapportés chez des patients traités par des
agonistes des récepteurs du GLP-1. La majorité des événements rapportés est
survenue chez des patients ayant présenté des nausées, des vomissements ou des
diarrhées entraînant une déplétion hydrique (voir rubrique Mises en garde
spéciales et précautions d'emploi).
Réactions
allergiques
Quelques cas
de réactions anaphylactiques associées à des symptômes tels qu'une hypotension,
des palpitations, une dyspnée et des œdèmes, ont été rapportés lors de la
commercialisation du liraglutide. Les réactions anaphylactiques peuvent
potentiellement engager le pronostic vital. Si une réaction anaphylactique est
suspectée, le liraglutide doit être arrêté et le traitement ne doit pas être
administré à nouveau (voir rubrique Contre-indications).
Réactions
au site d'injection
Des réactions
au site d'injection ont été rapportées chez des patients traités par Saxenda.
Ces réactions étaient habituellement légères et transitoires et disparaissaient
généralement lors de la poursuite du traitement.
Tachycardie
Lors des
essais cliniques, des cas de tachycardie ont été rapportés chez 0,6 % des
patients traités par Saxenda et chez 0,1 % des patients recevant un placebo. La
majorité des événements était d'intensité légère ou modérée. Ces événements
étaient isolés et la majorité d'entre eux s'est résolue lors de la poursuite du
traitement par Saxenda.
Amyloïdose cutanée
Une amyloïdose cutanée peut apparaitre au site d'injection (voir rubrique Posologie et mode d'administration).
Population pédiatrique
Lors d'un
essai clinique conduit chez des adolescents obèses âgés de 12 ans à moins de 18
ans, 125 patients ont été exposés à Saxenda pendant 56 semaines.
Dans
l'ensemble, la fréquence, la nature et la sévérité des effets indésirables chez
les adolescents obèses ont été comparables à celles observées dans la
population adulte. Les vomissements ont été deux fois plus fréquents chez les
adolescents que chez les adultes.
Le
pourcentage de patients rapportant au moins un épisode d'hypoglycémie
cliniquement significative a été plus élevé avec le liraglutide (1,6 %) qu'avec
le placebo (0,8 %). Aucun épisode d'hypoglycémie sévère n'est survenu pendant
l'essai.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via l'Agence nationale de sécurité du médicament et des produits de santé (ANSM) et le réseau des Centres Régionaux de Pharmacovigilance, site internet : https://signalement.social-sante.gouv.fr/.
SURVEILLANCE de la fréquence cardiaque à intervalles réguliers conformément à la pratique clinique habituelle.
INTERROMPRE le traitement après 12 semaines à la dose de 3,0 mg/jour si les patients n'ont pas perdu au moins 5 % de leur poids initial.
INFORMER les patients de 12 à 18 ans des symptômes caractéristiques et de la conduite à tenir en cas d'hypoglycémie.
ARRETER LE TRAITEMENT ET CONTACTER IMMEDIATEMENT UN MEDECIN en cas de :
- Douleur importante dans la partie supérieure du ventre, généralement
plus importante du côté droit sous les côtes, parfois jusque dans le
dos ou l' épaule droite.
- Problèmes pour respirer, un gonflement du visage et de la gorge et un rythme cardiaque rapide.
- Douleur intense et persistante dans l'abdomen (au niveau de
l'estomac) qui peut s'étendre au dos, associée à des nausées et des
vomissements
CONSULTER UN MEDECIN en cas de :
- Nodules thyroïdiens et augmentation de la taille de la glande thyroïdienne.
- Palpitations ou impression que le coeur s'emballe au repos.
BOIRE BEAUCOUP pour éviter la deshydratation.
INFORMER le médecin en cas d'intervention chirurgicale nécessitant une anesthésie (endormissement).
PRUDENCE en cas de conduite de véhicules ou d'utilisation de machines (vertiges).
Grossesse
Il existe des données limitées concernant l'utilisation du liraglutide chez la femme enceinte. Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique Données de sécurité préclinique). Le risque potentiel chez l'espèce humaine n'est pas connu.
Le liraglutide ne doit pas être utilisé pendant la grossesse. En cas de projet de grossesse ou en cas de grossesse, le traitement par le liraglutide devra être arrêté.
Allaitement
On ne sait pas si le liraglutide est excrété dans le lait maternel. Les études effectuées chez l'animal ont montré que le liraglutide et les métabolites à forte homologie structurelle étaient peu transférés dans le lait. Des études non cliniques réalisées chez de jeunes rats allaités ont mis en évidence un ralentissement de la croissance néonatale lié au traitement (voir rubrique Données de sécurité préclinique). En raison du manque d'expérience, Saxenda ne devra pas être utilisé pendant l'allaitement.
Fertilité
Hormis une légère diminution du nombre d'embryons vivants, les études effectuées chez l'animal n'ont pas mis en évidence d'effets délétères sur la fertilité (voir rubrique Données de sécurité préclinique).
In vitro, le liraglutide a montré un très faible potentiel d'interaction pharmacocinétique avec d'autres principes actifs se liant au cytochrome P450 (CYP) ou aux protéines plasmatiques.
Le léger ralentissement de la vidange gastrique observé avec le liraglutide est susceptible d'influencer l'absorption des médicaments administrés de façon concomitante par voie orale. Les études d'interaction n'ont pas mis en évidence de retard d'absorption cliniquement significatif et aucun ajustement de la dose n'est donc nécessaire.
Des études d'interaction ont été réalisées avec 1,8 mg de liraglutide. L'effet sur la vitesse de vidange gastrique était équivalent pour le liraglutide 1,8 mg et pour le liraglutide 3,0 mg (paracétamol ASC0-300min). Quelques patients traités par le liraglutide ont signalé au moins un épisode diarrhéique sévère. La diarrhée peut influencer l'absorption concomitante des médicaments pris par voie orale.
Warfarine et autres dérivés de la coumarine
Aucune étude d'interaction n'a été réalisée. Une interaction cliniquement significative avec des principes actifs peu solubles ou à marge thérapeutique étroite comme la warfarine ne peut être exclue. Lors de l'initiation du traitement par le liraglutide chez les patients sous warfarine ou autres dérivés de la coumarine, il est recommandé de surveiller plus fréquemment le Rapport Normalisé International (INR).
Paracétamol (acétaminophène)
Le liraglutide n'a pas modifié l'exposition totale au paracétamol après administration d'une dose unique de 1 000 mg. La Cmax du paracétamol a diminué de 31 % et le tmax médian a été retardé jusqu'à 15 min. Aucun ajustement de la dose n'est nécessaire en cas de prise concomitante de paracétamol.
Atorvastatine
Le liraglutide n'a pas modifié l'exposition totale à l'atorvastatine après administration d'une dose unique de 40 mg d'atorvastatine. Par conséquent, aucun ajustement de la dose d'atorvastatine n'est nécessaire en cas d'administration avec du liraglutide. Avec le liraglutide, la Cmax de l'atorvastatine a diminué de 38 % et le tmax médian a été retardé, passant de 1 h à 3 h.
Griséofulvine
Le liraglutide n'a pas modifié l'exposition totale à la griséofulvine après administration d'une dose unique de 500 mg de griséofulvine. La Cmax de la griséofulvine a augmenté de 37 % alors que le tmax médian n'a pas changé. Aucun ajustement de la dose de griséofulvine et des autres composés à solubilité faible et perméabilité élevée n'est nécessaire.
Digoxine
Après administration d'une dose unique de 1 mg de digoxine avec du liraglutide, l'ASC de la digoxine a été réduite de 16 % et la Cmax a diminué de 31 %. Le tmax médian de la digoxine a été retardé, passant de 1 h à 1,5 h. Ces résultats indiquent qu'aucun ajustement de la dose de la digoxine n'est nécessaire.
Lisinopril
Après administration d'une dose unique de 20 mg de lisinopril avec du liraglutide, l'ASC du lisinopril a été réduite de 15 % et la Cmax a diminué de 27 %. Avec le liraglutide, le tmax médian du lisinopril a été retardé, passant de 6 h à 8 h. Ces résultats indiquent qu'aucun ajustement de la dose du lisinopril n'est nécessaire.
Contraceptifs oraux
Après administration d'une dose unique d'un contraceptif oral, le liraglutide a diminué la Cmax de l'éthinylestradiol et du lévonorgestrel de 12 % et 13 % respectivement. Pour les deux composés, le tmax a été retardé de 1,5 h avec le liraglutide. Aucun effet cliniquement significatif sur l'exposition totale à l'éthinylestradiol et au lévonorgestrel n'a été observé. Il n'est pas attendu de modification de l'effet contraceptif lors d'une administration concomitante avec le liraglutide.
Population pédiatrique
Les études d'interaction n'ont été réalisées que chez l'adulte.
Posologie
Adultes
La dose
initiale est de 0,6 mg une fois par jour. La dose doit être augmentée jusqu'à
3,0 mg une fois par jour, par paliers de 0,6 mg espacés d'au moins une semaine,
pour améliorer la tolérance gastro- intestinale (voir tableau 2). Si
l'augmentation à la dose supérieure n'est pas tolérée pendant deux semaines
consécutives, l'arrêt du traitement doit être envisagé. Une dose quotidienne
supérieure à 3,0 mg n'est pas recommandée.
Tableau 2 Schéma d'augmentation de
la dose
| Dose | Semaines | |
|
Augmentation de la dose sur 4 semaines |
0,6 mg | 1 |
| 1,2 mg | 1 | |
| 1,8 mg | 1 | |
| 2,4 mg | 1 | |
| Dose d'entretien |
3,0 mg
|
|
Adolescents
(≥ 12 ans)
Pour les adolescents
âgés de 12 ans à moins de 18 ans, un schéma d'escalade de dose similaire à
celui utilisé pour les adultes doit être appliqué (voir tableau 2). La dose
doit être augmentée jusqu'à 3,0 mg (dose d'entretien) ou jusqu'à la dose
maximale tolérée. Une dose quotidienne supérieure à 3,0 mg n'est pas
recommandée.
Doses
oubliées
Si une dose
est oubliée dans les 12 heures qui suivent l'heure d'administration habituelle,
le patient doit prendre la dose oubliée dès que possible. S'il reste moins de
12 heures avant la dose suivante, le patient ne doit pas prendre la dose
oubliée et doit reprendre la dose suivante prévue selon le schéma
d'administration quotidienne. Une dose supplémentaire ou une dose plus
importante ne doit pas être prise pour compenser la dose oubliée.
Patients
ayant un diabète de type 2
Saxenda ne doit pas être
utilisé en association à un autre agoniste des récepteurs du GLP-1.
Au début du traitement par Saxenda, une réduction de la dose d'insuline ou des sécrétagogues de l'insuline (tels que les sulfamides hypoglycémiants) administrés de façon concomitante doit être envisagée afin de réduire le risque d'hypoglycémie. Une autosurveillance glycémique est nécessaire pour ajuster la dose d'insuline et des sécrétagogues de l'insuline (voir rubrique Mises en garde spéciales et précautions d'emploi).
Populations particulières
Sujets âgés
(≥ 65 ans)
Aucun ajustement de la dose n'est nécessaire en fonction de l'âge. L'expérience
clinique de ce traitement chez les patients ≥ 75 ans est limitée et l'utilisation
chez ces patients n'est pas recommandée (voir rubriques Mises en garde
spéciales et précautions d'emploi et Propriétés pharmacocinétiques).
Insuffisance
rénale
Aucun ajustement de la dose n'est nécessaire chez les patients présentant une
insuffisance rénale légère ou modérée (clairance de la créatinine ≥ 30
ml/min). Saxenda
n'est pas recommandé chez les patients présentant une insuffisance rénale
sévère (clairance de la créatinine < 30 ml/min), y compris les patients
présentant une insuffisance rénale terminale (voir rubriques Mises en garde spéciales
et précautions d'emploi, Effets indésirables et Propriétés
pharmacocinétiques).
Insuffisance
hépatique
Aucun ajustement de la dose n'est recommandé chez les patients présentant une
insuffisance hépatique légère ou modérée. Saxenda
n'est pas recommandé chez les patients présentant une insuffisance hépatique
sévère et doit être utilisé avec précaution chez les patients présentant une
insuffisance hépatique légère ou modérée (voir rubriques Mises en garde spéciales
et précautions d'emploi et Propriétés pharmacocinétiques).
Population
pédiatrique
Aucun ajustement de la dose n'est nécessaire chez les adolescents à partir de
12 ans.
La sécurité et l'efficacité de Saxenda chez les
enfants de moins de 12 ans n'ont pas été établies (voir rubrique Propriétés
pharmacodynamiques).
Mode d'administration
Saxenda doit être administré par voie sous-cutanée uniquement. Il ne doit pas être administré par voie intraveineuse ou intramusculaire.
Saxenda doit être administré une fois par jour, quel que soit le moment de la journée, indépendamment des repas. Il doit être injecté dans l'abdomen, la cuisse ou le haut du bras. Le site d'injection et le moment de l'injection peuvent être modifiés sans ajustement de la dose. Toutefois, il est préférable d'effectuer les injections de Saxenda à peu près au même moment de la journée, après avoir choisi l'heure la plus adaptée. Les sites d'injection doivent toujours être alternés afin de réduire le risque de dépôts amyloïdes au site d'injection (voir rubrique Effets indésirables).
Pour les instructions plus détaillées concernant l'administration, voir la rubrique Précautions particulières d'élimination et de manipulation.
Durée de conservation :
30 mois
Après la première utilisation : 1 mois
Précautions particulières de conservation :
À conserver au réfrigérateur (entre 2 °C et 8 °C).
Ne pas congeler.
À conserver à distance du compartiment de congélation.
Après la première utilisation : à conserver à une température ne dépassant pas 30 °C ou à conserver au réfrigérateur (entre 2 °C et 8 °C).
Conserver le capuchon sur le stylo, afin de le protéger de la lumière.
Certaines substances mélangées à Saxenda peuvent entraîner une dégradation du liraglutide. En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Des cas de surdosage, jusqu'à 72 mg (24 fois la dose recommandée pour
le contrôle du poids), ont été rapportés lors des essais cliniques et
après la commercialisation du liraglutide. Les événements rapportés
incluaient des nausées et des vomissements sévères ainsi qu'une
hypoglycémie sévère.
En cas de surdosage, un traitement symptomatique approprié doit être initié en fonction des signes cliniques et des symptômes du patient. Les signes cliniques de déshydratation doivent être recherchés chez le patient et la glycémie doit être surveillée.
Classe pharmacothérapeutique : Médicaments utilisés dans le diabète, analogue du glucagon-like peptide-1 (GLP-1). Code ATC : A10BJ02
Mécanisme d'action
Le liraglutide est un analogue du glucagon-like peptide-1 (GLP-1) humain acylé, dont la séquence d'acides aminés présente 97 % d'homologie avec le GLP-1 humain endogène. Le liraglutide se lie au récepteur du GLP-1 (GLP-1R) et l'active.
Le GLP-1 est un régulateur physiologique de l'appétit et de la prise alimentaire, mais son mécanisme d'action exact est encore peu connu. Lors des études effectuées chez l'animal, l'administration périphérique de liraglutide a entraîné une assimilation dans des régions cérébrales spécifiques impliquées dans la régulation de l'appétit. Le liraglutide, par l'activation spécifique du GLP-1R, a ainsi augmenté la satiété et diminué les principaux signaux de la faim, ce qui a entraîné une perte de poids.
Les récepteurs du GLP-1 sont également exprimés dans des régions spécifiques du cœur, du système vasculaire, du système immunitaire et des reins. Dans les modèles de souris ayant de l'athérosclérose, le liraglutide a permis de prévenir la progression de la plaque aortique et a réduit l'inflammation de la plaque. De plus, le liraglutide a eu un effet bénéfique sur les lipides plasmatiques. Le liraglutide n'a pas réduit la taille des plaques déjà établies.
Effets pharmacodynamiques
Le liraglutide entraîne une perte de poids chez l'homme essentiellement par la perte de masse adipeuse, la réduction relative de la graisse viscérale étant supérieure à celle de la graisse sous- cutanée. Le liraglutide régule l'appétit en augmentant la sensation de réplétion et de satiété tout en réduisant la sensation de faim et la consommation alimentaire préventive, ce qui diminue la prise alimentaire. Le liraglutide n'augmente pas la dépense énergétique comparativement au placebo.
Le liraglutide stimule la sécrétion d'insuline et réduit la sécrétion de glucagon de façon glucose- dépendante, ce qui abaisse la glycémie à jeun et la glycémie postprandiale. L'effet hypoglycémiant est plus prononcé chez les patients présentant un prédiabète et un diabète comparativement aux patients ayant une glycémie normale. Des essais cliniques suggèrent que le liraglutide améliore et prolonge la fonction bêta-cellulaire, selon le modèle HOMA-B et le rapport pro-insuline/insuline.
Efficacité et sécurité clinique
L'efficacité et la sécurité du liraglutide dans le contrôle du poids en association à un régime hypocalorique et une augmentation de l'activité physique ont été étudiées lors de quatre essais de phase 3 randomisés, en double aveugle, contrôlés versus placebo ayant inclus au total 5 358 patients adultes.
- Essai 1 (SCALE Obesity & Pre-Diabetes - 1839) : un total de 3 731 patients présentant
une obésité (IMC ≥ 30 kg/m2) ou un surpoids (IMC ≥ 27 kg/m2) avec
une dyslipidémie et/ou une hypertension ont été stratifiés selon le statut prédiabétique à la sélection et l'IMC à l'inclusion (≥
30 kg/m2 ou < 30 kg/m2). Les 3 731 patients ayant suivi les 56 semaines de
traitement ont tous été randomisés, et les 2 254 patients présentant un prédiabète à la sélection, et ayant suivi les 160 semaines
de traitement ont été randomisés. Les deux périodes de traitement ont été
suivies d'une période de suivi observationnel de 12 semaines qui correspond à
l'arrêt du médicament/placebo, ont été randomisés. Les interventions sur le
mode de vie, sous la forme d'un régime à faible valeur énergétique et
d'exercices physiques conseillés constituaient un traitement de base pour tous
les patients.
La semaine 56 de l'essai 1 a permis d'évaluer la perte de poids chez l'ensemble des 3 731 patients randomisés (dont 2 590 ont terminé l'étude).
La semaine 160 de l'essai 1 a permis d'évaluer le délai d'apparition du diabète de type 2 chez les 2 254 patients randomisés présentant un prédiabète (dont 1 128 ont terminé l'étude). - Essai 2 (SCALE Diabetes - 1922) : essai randomisé de 56 semaines évaluant la perte de poids chez 846 patients obèses ou en surpoids (dont 628 ont terminé l'étude) présentant un diabète de type 2 insuffisamment contrôlé (HbA1c comprise entre 7 et 10 %). Le traitement de fond au début de l'essai était soit un régime alimentaire et de l'activité physique seuls, ou en association avec un traitement par la metformine, un sulfamide hypoglycémiant ou une glitazone en monothérapie, soit une association de ces traitements.
- Essai 3 (SCALE Sleep Apnoea - 3970) : essai randomisé de 32 semaines évaluant la sévérité de l'apnée du sommeil et la perte de poids chez 359 patients obèses (dont 276 ont terminé l'étude) présentant une apnée obstructive du sommeil modérée ou sévère.
- Essai 4 (SCALE Maintenance - 1923) : essai randomisé de 56 semaines évaluant la perte de poids et la stabilisation du poids chez 422 patients obèses ou en surpoids (dont 305 ont terminé l'étude) présentant une hypertension artérielle ou une dyslipidémie après une perte de poids antérieure ≥ 5% obtenue par un régime hypocalorique.
Poids
Une
perte de poids plus importante a été obtenue avec le liraglutide
comparativement au placebo chez les patients obèses/en surpoids, dans tous les
groupes étudiés. Dans toutes les populations de l'essai, davantage de patients
ont obtenu une perte de poids ≥ 5 % et > 10 % avec le liraglutide qu'avec le placebo (tableaux 4-6). Dans la
partie 160 semaines de l'essai 1 la perte de poids est survenue principalement
pendant la première année et a été maintenue tout au long des 160 semaines.
Dans l'essai 4, davantage de patients ont réussi à stabiliser la perte de poids
obtenue avant le début du traitement par le liraglutide
que par le placebo (81,4 % et 48,9 % respectivement). Des données spécifiques
concernant la perte de poids, les répondeurs, la chronologie et la distribution
cumulative du changement du poids (%) dans les essais 1 à 4 sont présentées
dans les tableaux 4 à 8 et sur les figures 1, 2 et 3.
Réponse en perte de poids après 12 semaines de traitement
par le liraglutide (3,0 mg)
Les
répondeurs précoces étaient définis comme les patients ayant obtenu une perte
de poids ≥ 5 % après 12 semaines avec une dose thérapeutique de liraglutide (4 semaines d'augmentation de la dose et 12
semaines avec une dose thérapeutique). Dans la semaine 56 de l'essai 1, 67,5 %
des patients ont obtenu une perte de poids ≥ 5 % après 12 semaines. Dans
l'essai 2, 50,4 % des patients ont obtenu une perte de poids ≥ 5 % après
12 semaines. En cas de poursuite du traitement par le liraglutide,
il est prévu que 86,2 % de ces répondeurs précoces atteindront une perte de
poids ≥ 5 % et 51 % une perte de poids ≥ 10 % après un an de
traitement. La perte de poids moyenne prévue chez les répondeurs précoces qui
suivent le traitement pendant un an est de 11,2 % de leur poids à l'inclusion
(9,7 % chez les hommes et 11,6 % chez les femmes). Chez les patients ayant obtenu
une perte de poids < 5 % après 12 semaines de traitement par une dose
thérapeutique de liraglutide, la proportion de
patients n'atteignant pas une perte de poids ≥ 10 % après un an est de
93,4 %.
Contrôle glycémique
Le
traitement par le liraglutide a significativement
amélioré les paramètres glycémiques dans les sous- populations ayant une
glycémie normale, un prédiabète ou un diabète de type
2. Dans la semaine 56 de l'essai 1, le nombre de patients ayant développé un
diabète de type 2 était inférieur dans le groupe traité par le liraglutide que dans le groupe recevant un placebo (0,2 %
vs 1,1 %). Le nombre de patients prédiabétiques à
l'inclusion rétablis de leur prédiabète était plus
élevé dans le groupe traité par le liraglutide que
dans le groupe recevant un placebo (69,2 % vs 32,7 %). Dans la semaine 160 de
l'essai 1, le critère primaire d'efficacité correspondait à la proportion de
patients ayant présenté un diabète de type 2, le critère d'évaluation étant le
délai d'apparition du diabète. À la semaine 160, au cours du traitement, 3 % des
patients traités par Saxenda et 11% des patients
traités par placebo ont été diagnostiqués diabétiques de type 2. Le délai
d'apparition estimé du diabète de type 2 chez les patients traités par liraglutide 3,0 mg était 2,7 fois plus long (avec un
intervalle de confiance de 95 % [1,9 ; 3,9]), et le rapport de risque de
développer un diabète de type 2 était de 0,2 pour le liraglutide
versus placebo.
Facteurs de risque cardiométabolique
Le
traitement par le liraglutide a significativement
amélioré la pression artérielle systolique et le tour de taille comparativement
au placebo (tableaux 4, 5 et 6).
Index d'apnées-hypopnées (IAH)
Le
traitement par le liraglutide a significativement
réduit la sévérité de l'apnée obstructive du sommeil telle qu'évaluée par la
modification de l'IAH par rapport à l'inclusion comparativement au placebo
(tableau 7).
Tableau 4, essai 1 : changement du poids, de la glycémie et des paramètres cardiométaboliques à la semaine 56 par rapport à l'inclusion
|
Saxenda (n = 2 437) |
Placebo (n = 1 225) |
Saxenda vs. placebo |
|||
| Poids | |||||
| Inclusion, kg (ET) | 106,3 (21,2) | 106,3 (21,7) | - | ||
| Changement moyen à la semaine 56, % (IC 95 %) | -8,0 | -2,6 | -5,4** (-5,8; -5,0) | ||
| Changement moyen à la semaine 56, kg (IC 95 %) | -8,4 | -2,8 | -5,6** (-6,0; -5,1) | ||
| Proportion de patients ayant perdu ≥ 5 % de leur poids à la semaine 56, % (IC 95 %) | 63,5 | 26,6 | 4,8** (4,1; 5,6) | ||
| Proportion de patients ayant perdu > 10 % de leur poids à la semaine 56, % (IC 95 %) | 32,8 | 10,1 | 4,3** (3,5; 5,3) | ||
| Glycémie et facteurs cardiométaboliques | Inclusion | Changement | Inclusion | Changement | |
| HbA1c, % | 5,6 | -0,3 | 5,6 | -0,1 | -0,23** (-0,25; -0,21) |
| Glycémie à jeun, mmol/l | 5,3 | -0,4 | 5,3 | -0,01 | -0,38** (-0,42; -0,35) |
| Pression artérielle systolique, mmHg | 123,0 | -4,3 | 123,3 | -1,5 | -2,8** (-3,6; -2,1) |
| Pression artérielle diastolique, mmHg | 78,7 | -2,7 | 78,9 | -1,8 | -0,9* (-1,4; -0,4) |
| Tour de taille, cm | 115,0 | -8,2 | 114,5 | -4,0 | -4,2** (-4,7; -3,7) |
Analyse
complète. Pour le poids, l'HbA1c, la glycémie à jeun, la pression artérielle et
le tour de taille, les valeurs à l'inclusion sont des moyennes, les changements
à la semaine 56 par rapport à l'inclusion sont des moyennes estimées (moindres
carrés) et les différences de traitement à la semaine 56 sont des différences
de traitement estimées. Pour les proportions de patients ayant perdu ≥ 5
/ > 10 % de leur poids, les odds ratios estimés
sont présentés. Les valeurs post-inclusion manquantes ont été imputées à l'aide
de la dernière observation reportée.
* p < 0,05. ** p < 0,0001. IC = intervalle de confiance. ET = écart-type.
* p < 0,05. ** p < 0,0001. IC = intervalle de confiance. ET = écart-type.
Tableau 5, Essai 1 : changement du poids, de la glycémie et des paramètres cardiométaboliques à la semaine 160 par rapport à l'inclusion
|
Saxenda
(n = 1 472) |
Placebo (n = 738) |
Saxenda vs. placebo |
|||
| Poids | |||||
| Inclusion, kg (ET) | 107,6 (21,6) | 108,0 (21,8) | |||
|
Changement moyen à la semaine 160, % (IC 95 %) |
-6,2 | -1,8 | -4,3** (-4,9; -3,7) | ||
| Changement moyen à la semaine 160, kg (IC 95 %) | -6,5 | -2,0 | -4,6** (-5,3; -3,9) | ||
|
Proportion de patients ayant perdu ≥ 5 % de leur poids à la semaine 160, % (IC 95 %) |
49,6 | 23,4 | 3,2** (2,6; 3,9) | ||
| Proportion de patients ayant perdu > 10 % de leur poids à la semaine 160, % (IC 95 %) | 24,4 | 9,5 | 3,1** (2,3; 4,1) | ||
|
Glycémie et facteurs cardiométaboliques |
Inclusion | Changement | Inclusion | Changement | |
| HbA1c, % | 5,8 | -0,4 | 5,7 | -0,1 | -0,21** (-0,24; -0,18) |
| Glycémie à jeun, mmol/l | 5,5 | -0,4 | 5,5 | 0,04 | -0,4** (-0,5; -0,4) |
| Pression artérielle systolique, mmHg | 124,8 | -3,2 | 125,0 | -0,4 | -2,8** (-3,8; -1,8) |
| Pression artérielle diastolique, mmHg | 79,4 | -2,4 | 79,8 | -1,7 | -0,6 (-1,3; 0,1) |
| Tour de taille, cm | 116,6 | -6,9 | 116,7 | -3,4 | -3,5** (-4,2; -2,8) |
Analyse
complète. Pour le poids, l'HbA1c, la glycémie à jeun, la pression artérielle et
le tour de taille, les valeurs à l'inclusion sont des moyennes, les changements
à la semaine 160 par rapport à l'inclusion sont des moyennes estimées (moindres
carrés) et les différences de traitement à la semaine 160 sont des différences
de traitement estimées. Pour les proportions de patients ayant perdu ≥ 5
/ > 10 % de leur poids, les odds ratios estimés
sont présentés. Les valeurs post-inclusion manquantes ont été imputées à l'aide
de la dernière observation reportée. ** p < 0,0001. IC = intervalle de
confiance. ET = écart-type.

Figure 1 Changement du poids (%) par rapport à l'inclusion en fonction du temps dans l'essai 1 (0 -56 semaines)
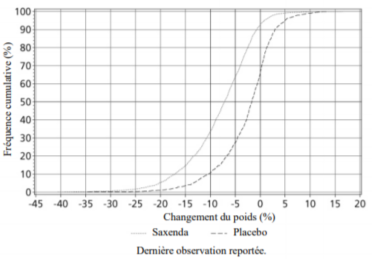
Figure 2 Distribution cumulative du changement du poids (%) après 56 semaines de traitement dans l'essai 1
Tableau 6, essai 2 : changement du poids, de la glycémie et des paramètres cardiométaboliques à la semaine 56 par rapport à l'inclusion
|
Saxenda (n = 412) |
Placebo (n = 211) |
Saxenda vs placebo |
|||
| Poids | |||||
| Inclusion, kg (ET) | 105,6 (21,9) | 106,7 (21,2) | - | ||
| Changement moyen à la semaine 56, % (IC 95 %) | -5,9 | -2,0 | -4,0** (-4,8; -3,1) | ||
| Changement moyen à la semaine 56, kg (IC 95 %) | -6,2 | -2,2 | -4,1** (-5,0; -3,1) | ||
| Proportion de patients ayant perdu ≥ 5 % de leur poids à la semaine 56, % (IC 95 %) | 49,8 | 13,5 | 6,4** (4,1; 10,0) | ||
| Proportion de patients ayant perdu > 10 % de leur poids à la semaine 56, % (IC 95 %) | 22,9 | 4,2 | 6,8** (3,4; 13,8) | ||
| Glycémie et facteurs cardiométaboliques | Inclusion | Changement | Inclusion | Changement | |
| HbA1c, % | 7,9 | -1,3 | 7,9 | -0,4 | -0,9** (-1,1; -0,8) |
| Glycémie à jeun, mmol/l | 8,8 | -1,9 | 8,6 | -0,1 | -1,8** (-2,1; -1,4) |
| Pression artérielle systolique, mmHg | 128,9 | -3,0 | 129,2 | -0,4 | -2,6* (-4,6; -0,6) |
| Pression artérielle diastolique, mmHg | 79,0 | -1,0 | 79,3 | -0,6 | -0,4 (-1,7; 1,0) |
| Tour de taille, cm | 118,1 | -6,0 | 117,3 | -2,8 | -3,2** (-4,2; -2,2) |
Analyse
complète. Pour le poids, l'HbA1c, la glycémie à jeun, la pression artérielle et
le tour de taille, les valeurs à l'inclusion sont des moyennes, les changements
à la semaine 56 par rapport à l'inclusion sont des moyennes estimées (moindres
carrés) et les différences de traitement à la semaine 56 sont des différences
de traitement estimées. Pour les proportions de patients ayant perdu ≥ 5
/ > 10 % de leur poids, les odds ratios estimés
sont présentés. Les valeurs post-inclusion manquantes ont été imputées à l'aide
de la dernière observation reportée. * p < 0,05. ** p < 0,0001. IC =
intervalle de confiance. ET = écart-type.
Tableau 7, essai 3 : changement du poids et de l'index d'apnées-hypopnées à la semaine 32 par rapport à l'inclusion
|
Saxenda (n = 180) |
Placebo (n = 179) |
Saxenda vs placebo |
|||
| Poids | |||||
| Inclusion, kg (ET) | 116,5 (23,0) | 118,7 (25,4) | - | ||
| Changement moyen à la semaine 32, % (IC 95 %) | -5,7 | -1,6 | -4,2** (-5,2; -3,1) | ||
| Changement moyen à la semaine 32, kg (IC 95 %) | -6,8 | -1,8 | -4,9** (-6,2; -3,7) | ||
| Proportion de patients ayant perdu ≥ 5 % de leur poids à la semaine 32, % (IC 95 %) | 46,4 | 18,1 | 3,9** (2,4; 6,4) | ||
| Proportion de patients ayant perdu > 10 % de leur poids à la semaine 32, % (IC 95 %) | 22,4 | 1,5 | 19,0** (5,7; 63,1) | ||
| Inclusion | Changement | Inclusion | Changement | ||
| Index d'apnées-hypopnées, événements/heure | 49,0 | -12,2 | 49,3 | -6,1 | -6,1* (-11,0; -1,2) |
* p < 0,05. ** p < 0,0001. IC = intervalle de confiance. ET = écart-type.
Tableau 8, essai 4 : changement du poids à la semaine 56 par rapport à l'inclusion
|
Saxenda
(n = 207) |
Placebo (n = 206) |
Saxenda vs placebo |
|
| Inclusion, kg (ET) | 100,7 (20,8) | 98,9 (21,2) | - |
| Changement moyen à la semaine 56, % (IC 95 %) | -6,3 | -0,2 | -6,1** (-7,5; -4,6) |
| Changement moyen à la semaine 56, kg (IC 95 %) | -6,0 | -0,2 | -5,9** (-7,3; -4,4) |
| Proportion de patients ayant perdu ≥ 5 % de leur poids à la semaine 56, % (IC 95 %) | 50,7 | 21,3 | 3,8** (2,4; 6,0) |
| Proportion de patients ayant perdu > 10 % de leur poids à la semaine 56, % (IC 95 %) | 27,4 | 6,8 | 5,1** (2,7; 9,7) |
Analyse
complète. Les valeurs à l'inclusion sont des moyennes, les changements à la
semaine 56 par rapport à l'inclusion sont des moyennes estimées (moindres
carrés) et les différences de traitement à la semaine 56 sont des différences
de traitement estimées. Pour les proportions de patients ayant perdu ≥ 5
/ > 10 % de leur poids, les odds ratios estimés
sont présentés. Les valeurs post-inclusion manquantes ont été imputées à l'aide
de la dernière observation reportée. ** p < 0,0001. IC = intervalle de
confiance. ET = écart-type.
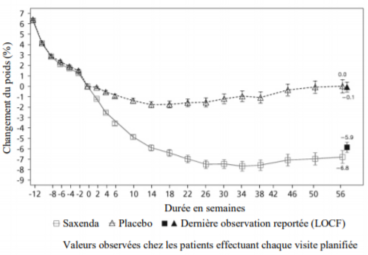
Figure 3 Changement du poids (%) par rapport à la randomisation (semaine 0) en fonction du temps dans l'essai 4
Avant la semaine 0, les patients avaient pour seul traitement un régime hypocalorique et de l'activité physique. Lors de la semaine 0, les patients ont été randomisés pour recevoir Saxenda ou le placebo.
Immunogénicité
Compte
tenu des propriétés potentiellement immunogènes des médicaments contenant des
protéines et des peptides, les patients traités par le liraglutide
peuvent développer des anticorps anti-liraglutide.
Lors des essais cliniques, 2,5 % des patients traités par le liraglutide ont développé des anticorps anti- liraglutide. L'apparition d'anticorps n'a pas été associée à une perte d'efficacité du liraglutide.
Évaluation cardiovasculaire
Les
événements indésirables cardiovasculaires majeurs ont été évalués par un groupe
d'experts indépendant externe et définis comme infarctus du myocarde non fatal,
accident vasculaire cérébral non fatal et décès cardiovasculaire. Dans
l'ensemble des essais cliniques à long terme menés avec Saxenda,
6 événements indésirables cardiovasculaires majeurs ont été observés chez les
patients traités par le liraglutide et 10 chez les
patients recevant un placebo. Le rapport de risque et l'IC 95 % est de 0,33
[0,12 ; 0,90] pour le liraglutide par rapport au
placebo. Une élévation moyenne du rythme cardiaque de 2,5 battements par minute
par rapport à l'inclusion (allant de 1,6 à 3,6 battements par minute d'un essai
à l'autre) a été observée avec le liraglutide dans
les essais cliniques de phase 3. Le rythme cardiaque a atteint son maximum
après environ 6 semaines. Le changement de rythme cardiaque était réversible à
l'arrêt du liraglutide (voir rubrique Mises en
garde spéciales et précautions d'emploi).
L'essai LEADER (Liraglutide Effect and Action in Diabetes Evaluation of Cardiovascular Outcomes Results) a inclus 9 340 patients avec un diabète de type 2 insuffisamment contrôlé. La grande majorité d'entre eux avaient une maladie cardiovasculaire établie. Les patients ont été randomisés pour recevoir soit le liraglutide à une dose quotidienne allant jusqu'à 1,8 mg (4 668) soit un placebo (4 672), tous deux en association avec une prise en charge standard.
La durée d'exposition était entre 3,5 ans et 5 ans. L'âge moyen était de 64 ans et l'IMC moyen était de 32,5 kg/m². La moyenne de l'HbA1c initiale était de 8,7 et s'est améliorée de 1,2 % chez les patients sous le liraglutide et de 0,8 % chez les patients sous placebo après 3 ans. Le critère primaire était le délai de survenue depuis la randomisation du premier événement cardiovasculaire majeur (MACE) : mortalité cardiovasculaire, infarctus du myocarde non fatal ou accident vasculaire cérébral non fatal.
Le liraglutide a significativement diminué le taux d'événements cardiovasculaires majeurs (critère primaire, MACE) vs. placebo (3,41 vs. 3,90 par 100 patients-années d'observation dans le groupe liraglutide et placebo respectivement) avec une réduction du risque de 13 %, HR 0,87, [0,78 ; 0,97] [IC 95 %] (p = 0,005) (voir figure 4).
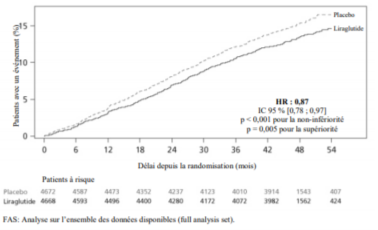
Figure 4 Kaplan Meier du délai de survenue du premier MACE - Analyse de la population FAS
Population pédiatrique
L'Agence
européenne des médicaments a différé l'obligation de soumettre les résultats
d'études réalisées avec Saxenda dans un ou plusieurs
sous-groupes de la population pédiatrique dans le traitement de l'obésité (voir
rubrique Posologie et mode d'administration pour les informations
concernant l'usage pédiatrique).
Lors
d'un essai en double aveugle comparant l'efficacité et la sécurité de Saxenda versus placebo sur la perte de poids chez des
patients adolescents obèses âgés de 12 ans et plus, Saxenda
a été supérieur au placebo en termes de perte de poids (évaluée par le score
d'écart-type [SET] de l'IMC) après 56 semaines de traitement (tableau 9).
Une
proportion plus importante des patients a obtenu des réductions de l'IMC ≥
5 % et ≥ 10 % avec le liraglutide versus
placebo ainsi que des réductions plus importantes des valeurs moyennes de l'IMC
et du poids (tableau 9). Après une période de suivi de
26 semaines sans administration du produit à l'étude, une reprise de poids a
été observée avec le liraglutide versus placebo
(tableau 9).
Tableau 9 Essai 4180 : Changement du poids et de l'IMC à la semaine 56 par rapport à l'inclusion et changement du SET de l'IMC entre la semaine 56 et la semaine 82
|
Saxenda (N = 125) |
Placebo (N = 126) |
Saxenda versus placebo |
|
| SET de l'IMC | |||
| Inclusion, SET de l'IMC (ET) | 3,14 (0,65) | 3,20 (0,77) | |
| Changement moyen à la semaine 56 (IC 95 %) | -0,23 | 0,00 | -0,22* (-0,37; -0,08) |
| Semaine 56, SET de l'IMC (ET) | 2,88 (0,94) | 3,14 (0,98) | |
| Changement moyen entre la semaine 56 et la semaine 82, SET de l'IMC (IC 95 %) | 0,22 | 0,07 | 0,15** (0,07; 0,23) |
| Poids | |||
| Inclusion, kg (ET) | 99,3 (19,7) | 102,2 (21,6) | - |
| Changement moyen à la semaine 56, % (IC 95 %) | -2,65 | 2,37 | -5,01** (-7,63; -2,39) |
| Changement moyen à la semaine 56, kg (IC 95 %) | -2,26 | 2,25 | -4,50** (-7,17; -1,84) |
| IMC | |||
| Inclusion, kg/m² (ET) | 35,3 (5,1) | 35,8 (5,7) | - |
| Changement moyen à la semaine 56, kg/m² (IC 95 %) | -1,39 | 0,19 | -1,58** (-2,47; -0,69) |
| Proportion de patients avec une réduction ≥ 5 % de l'IMC à la semaine 56 par rapport à l'inclusion, % (IC 95 %) | 43,25 | 18,73 | 3,31** (1,78; 6,16) |
| Proportion de patients avec une réduction ≥ 10 % de l'IMC à la semaine 56 par rapport à l'inclusion, % (IC 95 %) | 26,08 | 8,11 | 4,00** (1,81 ; 8,83) |
Analyse
complète. Pour le SET de l'IMC, le poids et l'IMC, les valeurs à l'inclusion
sont des moyennes, les changements à la semaine 56 par rapport à l'inclusion
sont des moyennes estimées (moindres carrés) et les différences de traitement à
la semaine 56 sont des différences de traitement estimées. Pour le SET de
l'IMC, les valeurs à la semaine 56 sont des moyennes, les changements entre la
semaine 56 et la semaine 82 sont des moyennes estimées (moindres carrés) et les
différences de traitement à la semaine 82 sont des différences de traitement
estimées. Pour les proportions de patients ayant perdu ≥ 5 %/≥ 10 %
de leur IMC à l'inclusion, les odds ratios estimés
sont présentés. Les observations manquantes ont été imputées à partir du groupe
placebo en utilisant la méthode d'imputation multiple (x 100) par saut de
référence.
*p < 0,01, **p < 0,001. IC = intervalle de confiance. ET = écart-type.
*p < 0,01, **p < 0,001. IC = intervalle de confiance. ET = écart-type.
Sur
la base de la tolérance, 103 patients (82,4 %) sont passés et restés à la dose
de 3,0 mg, 11 patients (8,8 %) sont passés et restés à la dose de 2,4 mg, 4
patients (3,2 %) sont passés et restés à la dose de 1,8 mg, 4 patients (3,2 %)
sont passés et restés à la dose de 1,2 mg et 3 patients (2,4 %) sont restés à
la dose de 0,6 mg.
Aucun
effet sur la croissance ou le développement pubertaire n'a été observé après 56
semaines de traitement.
Une
étude menée 16 semaines en double aveugle et 36 semaines en ouvert a été
réalisée afin d'évaluer l'efficacité et la sécurité de Saxenda
chez des enfants adolescents atteints du syndrome de Prader-Willi
et présentant une obésité. L'étude a inclus 32 patients âgés de 12 à < 18
ans (partie A) et 24 patients âgés de 6 à < 12 ans (partie B). Les patients
ont été randomisés en 2:1 pour recevoir Saxenda ou un placebo. Les patients ayant un poids corporel
inférieur à 45 kg ont débuté l'escalade de dose à une dose plus faible ; 0,3 mg
au lieu de 0,6 mg, jusqu'à une dose maximale de 2,4 mg.
La
différence estimée entre les traitements par le SET de l'IMC (moyenne), à 16
semaines (partie A : - 0,20 vs -0,13 ; partie B : -0,50 vs -0,44) et à 52
semaines (partie A : -0,31 vs -0,17 ; partie B : -0,73 vs -0,67) était
similaire avec Saxenda et le placebo.
Aucun
problème additionnel de sécurité n'a été observé au cours de l'essai.
Absorption
L'absorption du liraglutide administré par voie sous-cutanée était lente. La concentration maximale était atteinte environ 11 heures après l'injection. La concentration moyenne du liraglutide à l'état d'équilibre (ASCτ/24) était d'environ 31 nmol/l chez les patients obèses (IMC de 30 à 40 kg/m2) après l'administration de 3 mg de liraglutide. L'exposition au liraglutide a augmenté proportionnellement à la dose. La biodisponibilité absolue du liraglutide après administration sous-cutanée est d'environ 55 %.
Distribution
Le volume de distribution apparent moyen après administration sous-cutanée est de 20 à 25 l (chez une personne pesant environ 100 kg). Le liraglutide se lie largement aux protéines plasmatiques (> 98 %).
Biotransformation
Dans les 24 heures suivant l'administration d'une dose unique de [3H]-liraglutide à des sujets sains, le principal composant plasmatique était le liraglutide intact. Deux métabolites plasmatiques mineurs ont été détectés (≤ 9 % et ≤ 5 % de la radioactivité plasmatique totale).
Élimination
Le liraglutide est métabolisé par voie endogène de la même manière que les grosses protéines et aucun organe en particulier n'a été identifié comme voie d'élimination principale. Après administration d'une dose de [3H]-liraglutide, le liraglutide intact n'a été détecté ni dans les urines ni dans les fèces. Seule une proportion minime de la radioactivité administrée a été excrétée dans les urines ou dans les fèces sous forme de métabolites issus du liraglutide (6 % et 5 % respectivement). La radioactivité urinaire et fécale a été principalement excrétée pendant les 6 à 8 premiers jours respectivement sous la forme de trois métabolites mineurs.
Après administration sous-cutanée de liraglutide, la clairance moyenne est d'environ 0,9 à 1,4 l/h avec une demi-vie d'élimination d'environ 13 heures.
Populations particulières
Patients âgés
L'âge n'a aucun effet cliniquement significatif sur la pharmacocinétique du liraglutide d'après les résultats d'une analyse des données pharmacocinétiques de population chez des patients obèses ou en surpoids de 18 à 82 ans. Aucun ajustement posologique n'est nécessaire en fonction de l'âge.
Sexe
D'après les résultats des analyses pharmacocinétiques de population, les femmes présentent une clairance du liraglutide ajustée selon le poids de 24 % inférieure à celle des hommes. D'après les données de réponse à l'exposition, aucun ajustement posologique n'est nécessaire en fonction du sexe.
Origine ethnique
L'origine ethnique n'a aucun effet cliniquement significatif sur la pharmacocinétique du liraglutide d'après les résultats d'une analyse pharmacocinétique de population incluant des patients obèses ou en surpoids d'ethnie blanche, noire, asiatique et hispanique/non hispanique.
Poids
L'exposition au liraglutide diminue quand le poids à l'inclusion augmente. La dose quotidienne de 3,0 mg de liraglutide assurait une exposition systémique adéquate chez des patients d'un poids compris entre 60 et 234 kg lors de l'évaluation de la réponse à l'exposition dans les essais cliniques. L'exposition au liraglutide n'a pas été étudiée chez des patients d'un poids > 234 kg.
Insuffisance hépatique
La pharmacocinétique du liraglutide a été évaluée chez des patients présentant un degré variable d'insuffisance hépatique dans un essai en dose unique (0,75 mg). L'exposition au liraglutide a diminué de 13 à 23 % chez les patients qui présentaient une insuffisance hépatique légère à modérée par rapport aux sujets sains. L'exposition était significativement diminuée (44 %) chez les patients présentant une insuffisance hépatique sévère (score de Child Pugh > 9).
Insuffisance rénale
L'exposition au liraglutide s'est révélée plus faible chez les patients qui présentaient une insuffisance rénale par rapport aux sujets ayant une fonction rénale normale dans un essai en dose unique (0,75 mg). L'exposition au liraglutide a diminué de 33 %, 14 %, 27 % et 26 % chez les patients présentant respectivement une insuffisance rénale légère (clairance de la créatinine Clcr de 50 à 80 ml/min), modérée (Clcr de 30 à 50 ml/min), sévère (Clcr < 30 ml/min) et en insuffisance rénale terminale nécessitant une dialyse.
Population pédiatrique
Les propriétés pharmacocinétiques pour le liraglutide 3,0 mg ont été évaluées dans des études cliniques chez les patients adolescents obèses âgés de 12 à moins de 18 ans (134 patients, poids corporel allant de 62 à 178 kg). L'exposition au liraglutide chez les adolescents (âgés de 12 à moins de 18 ans) était similaire à celle observée chez les adultes obèses.
Les
propriétés pharmacocinétiques ont également été évaluées dans une étude de
pharmacologie clinique dans la population pédiatrique obèse âgée de 7 à 11 ans
(13 patients, poids corporel allant de 54 à 87 kg) respectivement.
L'exposition au liraglutide 3,0 mg était similaire
chez les enfants âgés de 7 à 11 ans, les adolescents et les adultes obèses
après ajustement sur les différences de poids corporel.
Saxenda n'a aucun effet ou qu'un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines. Cependant, des vertiges peuvent apparaître principalement au cours des 3 premiers mois de traitement avec Saxenda. La conduite ou l'utilisation de machines doivent être réalisées avec prudence si des vertiges se manifestent.
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration répétée ou génotoxicité n'ont pas révélé de risque particulier pour l'homme.
Des tumeurs non létales des cellules C de la thyroïde ont été observées lors d'études de carcinogénicité sur deux ans chez le rat et la souris. Chez le rat, aucune dose sans effet nocif observé (DSENO) n'a été observée. Ces tumeurs n'ont pas été observées chez des singes traités pendant 20 mois. Ces résultats décrits chez les rongeurs sont dus à un mécanisme non génotoxique, spécifique, médié par les récepteurs du GLP-1, auquel les rongeurs sont particulièrement sensibles. La pertinence de ces résultats pour l'homme est probablement faible mais ne peut pas être complètement exclue. Aucun autre type de tumeurs liées au traitement n'a été identifié.
Les études effectuées chez l'animal n'ont mis en évidence aucun effet délétère direct sur la fertilité mais une légère augmentation des morts embryonnaires précoces a été observée à la dose la plus élevée. L'administration de liraglutide en milieu de gestation a entraîné une perte de poids maternelle et une diminution de la croissance fœtale, avec des effets ambigus sur la cage thoracique chez le rat et une modification du squelette chez le lapin. Chez les rats exposés au liraglutide, on a observé un ralentissement de la croissance néonatale qui a persisté après le sevrage chez le groupe recevant des doses élevées. Il n'est pas établi si le retard de croissance des jeunes rats est imputable à une consommation de lait réduite due à un effet direct du GLP-1 ou à une baisse de la production de lait maternel induite par une réduction de l'apport calorique.
Chez les rats juvéniles, le liraglutide a entraîné un retard de la maturation sexuelle aussi bien chez les mâles que chez les femelles à des expositions cliniques pertinentes. Ces retards n'ont pas eu d'impact sur la fertilité et la capacité de reproduction de chacun des deux sexes ou sur la capacité des femelles à maintenir une gestation.
La solution ne doit pas
être utilisée si elle n'est pas limpide et incolore ou presque
incolore.
Saxenda ne doit pas être utilisé s'il a été congelé.
Le stylo est conçu pour être utilisé avec les aiguilles NovoFine ou NovoTwist à usage unique d'une longueur maximale de 8 mm et d'un diamètre minimal de 32 G.
Les aiguilles ne sont pas incluses.
Le patient doit être averti du fait qu'il doit jeter l'aiguille d'injection après chaque injection et conserver le stylo sans aiguille d'injection attachée. Ceci prévient les risques de contamination, d'infection et de fuite. Cela garantit également la précision du dosage.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
- Prescription initiale réservée aux spécialistes en
endocrinologie - diabétologie - nutrition ou aux médecins compétents en nutrition.
- Renouvellement non restreint.
Solution injectable.
Solution isotonique, incolore ou presque incolore et limpide ; pH = 8,15.
Cartouche (verre de type 1) munie d'un piston (bromobutyle) et d'une feuille en caoutchouc stratifiée (bromobutyle/polyisoprène) contenue dans un stylo prérempli multidose jetable en polypropylène, polyacétal, polycarbonate et acrylonitrile butadiène styrène.
Chaque stylo contient 3 ml de solution et peut délivrer des doses de 0,6 mg, 1,2 mg, 1,8 mg, 2,4 mg et 3,0 mg.
Boîte de 3 stylos préremplis.
1 ml de solution contient 6 mg de liraglutide*. Un stylo prérempli contient 18 mg de liraglutide dans 3 ml.
* peptide analogue au glucagon-1 humain (GLP-1) produit par la technique de l'ADN recombinant sur Saccharomyces cerevisiae.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Phosphate disodique dihydraté
Propylène glycol
Phénol
Acide chlorhydrique (pour ajustement du pH)
Hydroxyde de sodium (pour ajustement du pH)
Eau pour préparations injectables
Novo Nordisk
12 Cours Michelet
Carré Michelet
92800
Puteaux
Téléphone : 01 41 97 66 00
Information médicale
Tél
:
Ou 01 41
97 65 00 (Service et appel gratuits)
Fax : 
Email :
infomed@novonordisk.com
Site web : http://www.novonordisk.fr