
LOJUXTA 5 mg, gélule, boîte de 1 flacon de 28
Dernière révision : 27/01/2025
Taux de TVA : 2.1%
Prix de vente : 20 610,06 €
Taux remboursement SS : 65%
Base remboursement SS : 20 610,06 €
Laboratoire exploitant : CHIESI
Source :

Lojuxta est indiqué en complément d'un régime alimentaire pauvre en graisses et d'autres médicaments hypolipémiants, avec ou sans aphérèse des lipoprotéines de basse densité (LDL), chez des patients adultes présentant une hypercholestérolémie familiale homozygote (HFHo).
L'HFHo doit être confirmée par un test génétique dans toute la mesure du possible. D'autres formes d'hyperlipoprotéinémies primaires et les causes secondaires d'hypercholestérolémie (syndrome néphrotique, hypothyroïdisme, par exemple) doivent être exclues.
- Hypersensibilité au principe actif ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
- Patients présentant une insuffisance hépatique modérée ou sévère et patients dont les tests de la fonction hépatique sont anormaux de façon prolongée et inexpliquée (voir rubrique Posologie et mode d'administration).
- Patients souffrant d'une maladie intestinale importante ou chronique connue, par exemple une maladie intestinale inflammatoire ou de malabsorption.
- Administration concomitante de plus de 40 mg de simvastatine (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
- Utilisation concomitante de Lojuxta et d'inhibiteurs puissants ou modérés du CYP3A4 (p. ex. antifongiques azolés tels que l'itraconazole, le fluconazole, le kétoconazole, le voriconazole, le posaconazole ; antibiotiques macrolides, comme l'érythromycine ou la clarithromycine ; antibiotiques kétolides tels que la télithromycine ; inhibiteurs des protéases du VIH ; les bloqueurs de canaux calciques diltiazem et vérapamil, ainsi que le médicament anti-arythmique dronédarone [voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions]).
- Grossesse (voir rubrique Fertilité, grossesse et allaitement).
Anomalies des enzymes hépatiques
Le lomitapide peut provoquer des augmentations des taux des enzymes hépatiques alanine aminotransférase [ALAT] et aspartate aminotransférase [ASAT] et une stéatose hépatique (voir rubrique Propriétés pharmacodynamiques). Il n'y a pas eu d'augmentation concomitante ou subséquente cliniquement significative de la bilirubine sérique, du rapport international normalisé (INR, International Normalised Ratio), ni des phosphatases alcalines. Il n'est pas connu dans quelle mesure la stéatose hépatique associée au lomitapide favorise l'augmentation des taux d'aminotransférases. Les modifications des enzymes hépatiques peuvent se produire à n'importe quel moment pendant le traitement, mais se sont produites le plus souvent lors de l'augmentation progressive de la dose.
Bien que des cas de dysfonctionnement du foie (taux élevés des aminotransférases associés à une augmentation de la bilirubine ou de l'INR) ou d'insuffisance hépatique n'aient pas été rapportés, le lomitapide pourrait induire une stéato-hépatite, pouvant évoluer en cirrhose sur plusieurs années. Les études cliniques étayant la sécurité et l'efficacité du lomitapide dans l'HFHo n'auraient probablement pas permis de détecter ce résultat indésirable, en raison de leur taille et de leur durée.
Surveillance des tests de la fonction hépatique
Les taux des enzymes ALAT, ASAT, phosphatases alcalines, de la bilirubine totale, de la gamma-glutamyl transférase (gamma-GT) et de l'albumine sérique doivent être mesurés avant de commencer le traitement par Lojuxta. Ce médicament est contre-indiqué chez les patients présentant une insuffisance hépatique modérée ou sévère et les patients dont les tests de la fonction hépatique sont anormaux de façon prolongée et inexpliquée. Si les résultats des tests hépatiques avant traitement sont anormaux, l'instauration du traitement par ce médicament ne doit être envisagée qu'après une investigation appropriée réalisée par un hépatologue et seulement lorsque les anomalies avant traitement sont expliquées ou résolues.
Pendant la première année de traitement, il faut mesurer les valeurs des tests hépatiques (au minimum les taux d'ALAT et d'ASAT), avant chaque augmentation de dose ou une fois par mois, selon l'échéance qui se présente en premier. Après la première année, ces analyses doivent être réalisées au moins tous les 3 mois et avant toute augmentation de dose. Il convient de réduire la dose de Lojuxta si des augmentations des taux d'aminotransférases sont observées et d'interrompre le traitement en cas d'augmentations prolongées ou cliniquement significatives (voir le Tableau 1).
Modification de la dose en raison d'augmentations des taux d'aminotransférases hépatiques
Le Tableau 1 résume les recommandations à suivre pour l'ajustement de la dose et la surveillance chez les patients qui développent des taux élevés d'aminotransférases pendant le traitement par Lojuxta.
Tableau 1 : Ajustement de la dose et surveillance des patients présentant des taux élevés d'aminotransférases
|
ALAT ou ASAT |
Recommandations pour le traitement et le suivi des patients* |
|
≥ à 3 fois et < à 5 fois la limite supérieure de la normale (LSN) |
• Confirmer l'augmentation par une nouvelle mesure dans un délai d'une semaine. • En cas de confirmation, réduire la dose et faire réaliser des tests complémentaires de la fonction hépatique s'ils n'ont pas déjà été effectués (tels que phosphatases alcalines, bilirubine totale et INR). • Renouveler les analyses toutes les semaines et interrompre la prise du médicament s'il y a des signes de fonctionnement anormal du foie (augmentation de la bilirubine totale ou de l'INR), si les taux d'aminotransférases dépassent 5 fois la LSN ou s'ils ne redescendent pas en dessous de 3 fois la LSN dans un délai d'environ 4 semaines. Adresser les patients présentant des taux élevés d'aminotransférases supérieurs à 3 fois la LSN de façon prolongée à un hépatologue pour des investigations complémentaires. • En cas de reprise du traitement par Lojuxta après un retour des taux d'aminotransférases en dessous de 3 fois la LSN, envisager de réduire la dose et surveiller les tests de la fonction hépatique plus souvent. |
|
≥ à 5 fois la LSN |
• Interrompre la prise du médicament et faire réaliser des tests complémentaires de la fonction hépatique s'ils n'ont pas déjà été effectués (tels que phosphatases alcalines, bilirubine totale et INR). Si les taux d'aminotransférases ne reviennent pas en dessous de 3 fois la LSN dans un délai d'environ 4 semaines, adresser le patient à un hépatologue pour des investigations complémentaires. • En cas de reprise du traitement par Lojuxta après un retour des taux d'aminotransférases en dessous de 3 fois la LSN, réduire la dose et surveiller les tests de la fonction hépatique plus souvent. |
*Recommandations pour une LSN d'environ 30 à 40 unités internationales/litre.
Si les augmentations des taux d'aminotransférases s'accompagnent de symptômes cliniques d'atteinte hépatique (tels que nausées, vomissements, douleurs abdominales, fièvre, jaunisse, léthargie, symptômes grippaux), d'augmentations de la bilirubine supérieures ou égales à 2 fois la LSN ou d'une maladie hépatique évolutive, le traitement par Lojuxta doit être interrompu et le patient doit être adressé à un hépatologue pour des investigations complémentaires.
La réintroduction du traitement peut être envisagée si les bénéfices sont estimés supérieurs aux risques associés à une atteinte hépatique potentielle.
Stéatose hépatique et risque de maladie hépatique évolutive
En cohérence avec le mécanisme d'action du lomitapide, la majorité des patients traités ont présenté une augmentation des graisses hépatiques. Dans une étude de phase 3, en ouvert, 18 patients sur 23 présentant une HFHo ont développé une stéatose hépatique (graisses hépatiques > 5,56 %), d'après des mesures par spectroscopie par résonance magnétique nucléaire (RMN) (voir rubrique Propriétés pharmacodynamiques). L'augmentation absolue médiane des graisses hépatiques a été de 6 % après 26 et 78 semaines de traitement, par rapport à 1 % avant traitement, mesurée par spectroscopie par RMN. La stéatose hépatique est un facteur de risque pour des maladies hépatiques évolutives, notamment la stéato-hépatite et la cirrhose. Les conséquences à long terme de la stéatose hépatique associée au traitement par lomitapide ne sont pas connues. Des données cliniques suggèrent que l'accumulation de graisses dans le foie est réversible après l'arrêt du traitement par Lojuxta, mais on ne sait pas s'il subsiste des séquelles histologiques, en particulier après une utilisation de longue durée.
Surveillance pour détecter des signes de maladie hépatique évolutive
Un dépistage régulier d'une stéato-hépatite ou d'une fibrose doit être effectué avant traitement, puis annuellement, à l'aide des techniques d'imagerie et des biomarqueurs suivants :
- imagerie visant à déterminer l'élasticité des tissus, par exemple FibroScan, imagerie par impulsionde force de radiation acoustique (ARFI, acoustic radiation force impulse), ou élastographie par résonance magnétique (RM)
- mesure des gamma-GT et de l'albumine sérique pour détecter de possibles lésions hépatiques
- mesure d'au moins un des marqueurs dans chacune des catégories suivantes :
- protéine C réactive de haute sensibilité (PCR-hs), vitesse de sédimentation des érythrocytes (VSE), cytokératine 18 (fragment CK-18), NASH Test (recherche d'une inflammation du foie)
- score ELF (Enhanced Liver Fibrosis), FibroMètre, rapport ASAT/ALAT, score Fib-4, Fibrotest (fibrose hépatique)
La réalisation de ces tests et leur interprétation devront faire intervenir le médecin traitant les lipides et l'hépatologue. Chez les patients dont les résultats suggèrent la présence d'une stéato-hépatite ou d'une fibrose une biopsie hépatique doit être envisagée.
Si un patient souffre d'une stéato-hépatite ou d'une fibrose confirmée par biopsie, le rapport bénéfice/risque doit être réévalué et le traitement doit être arrêté si nécessaire.
Déshydratation
Des cas de déshydratation et d'hospitalisation ont été rapportés chez des patients traités par le lomitapide depuis sa commercialisation. Les patients traités par le lomitapide doivent être avertis du risque potentiel de déshydratation liée aux réactions indésirables gastro-intestinales et doivent prendre des précautions pour éviter une déplétion hydrique.
Utilisation concomitante d'inhibiteurs du CYP3A4
Le lomitapide s'avère être un substrat sensible pour le CYP3A4. Les inhibiteurs du CYP3A4 augmentent l'exposition au lomitapide, les inhibiteurs puissants induisant une augmentation de l'exposition d'un facteur 27 environ. L'utilisation concomitante d'inhibiteurs modérés ou puissants du CYP3A4 et de Lojuxta est contre-indiquée (voir rubrique Contre-indications). Dans les études cliniques menées avec le lomitapide, un patient présentant une HFHo a développé une augmentation marquée des taux d'aminotransférases (de 24 fois la LSN pour l'ALAT, de 13 fois pour l'ASAT) dans les jours qui ont suivi l'instauration d'un traitement par la clarithromycine, un inhibiteur puissant du CYP3A4. Si le traitement par des inhibiteurs modérés ou puissants du CYP3A4 est inévitable, Lojuxta doit être arrêté pendant la durée du traitement.
Les inhibiteurs faibles du CYP3A4 sont susceptibles d'augmenter l'exposition au lomitapide lorsqu'ils sont administrés de façon concomitante. En cas d'administration avec l'atorvastatine, la dose de Lojuxta doit soit être prise à 12 heures d'intervalle soit être diminuée de moitié (voir rubrique Posologie et mode d'administration). La dose de Lojuxta doit être administrée à 12 heures d'intervalle de n'importe quel autre inhibiteur faible du CYP3A4.
Utilisation concomitante d'inducteurs du CYP3A4
Les médicaments inducteurs du CYP3A4 sont susceptibles d'augmenter la vitesse et l'intensité du métabolisme du lomitapide. Les inducteurs du CYP3A4 exercent leur effet de manière temps-dépendante et peuvent nécessiter au moins 2 semaines pour atteindre leur effet maximal après leur introduction. Inversement, lors de l'arrêt de ces médicaments, la baisse de l'induction du CYP3A4 peut prendre au moins 2 semaines.
L'administration concomitante d'un inducteur du CYP3A4 peut entraîner une diminution de l'effet du lomitapide. Tout effet sur l'efficacité est susceptible d'être variable. Lors de l'administration concomitante d'inducteurs du CYP3A4 (par ex. aminoglutéthimide, nafcilline, inhibiteurs non nucléosidiques de la transcriptase inverse, phénobarbital, rifampicine, carbamazépine, pioglitazone, glucocorticoïdes, modafinil et phénytoïne) et de Lojuxta, la possibilité d'une interaction médicamenteuse susceptible d'agir sur l'efficacité doit être prise en compte. L'utilisation de millepertuis avec Lojuxta doit être évitée.
Si une utilisation chronique de l'inducteur du CYP3A4 est envisagée, il est recommandé d'augmenter la fréquence des mesures du C-LDL et d'envisager d'augmenter la dose de Lojuxta pour assurer le maintien du niveau d'efficacité souhaité. Lors de la suppression d'un inducteur du CYP3A4, la possibilité d'une exposition accrue doit être prise en compte et il peut être nécessaire de réduire la dose de Lojuxta.
Utilisation concomitante d'inhibiteurs de l'HMG-CoA (« statines »)
Le lomitapide augmente les concentrations plasmatiques des statines. Les patients recevant Lojuxta en complément d'un traitement par une statine doivent être surveillés pour détecter des événements indésirables associés à l'utilisation de fortes doses de statines. Ces dernières induisent quelquefois une myopathie. Dans de rares cas, la myopathie peut prendre la forme d'une rhabdomyolyse, avec ou sans insuffisance rénale aiguë secondaire à une myoglobinurie, et peut conduire au décès. Tous les patients recevant du lomitapide en même temps qu'une statine doivent être avertis du risque accru potentiel de myopathie et il convient de leur demander de signaler rapidement toute douleur, sensibilité ou faiblesse musculaire inexpliquée. Des doses de simvastatine supérieures à 40 mg ne doivent pas être utilisées avec Lojuxta (voir rubrique Contre-indications).
Jus de pamplemousse
Le jus de pamplemousse doit être supprimé du régime alimentaire pendant le traitement par Lojuxta.
Risque d'anticoagulation suprathérapeutique ou subthérapeutique avec des anticoagulants coumariniques
Le lomitapide augmente les concentrations plasmatiques de warfarine. Des augmentations de la dose de Lojuxta peuvent entraîner une anticoagulation suprathérapeutique et des diminutions de la dose peuvent conduire à une anticoagulation subthérapeutique. La difficulté à contrôler l'INR a contribué à l'arrêt précoce de l'étude de phase 3 chez un des cinq patients prenant simultanément de la warfarine. Les patients prenant de la warfarine doivent être soumis à une surveillance régulière de l'INR, en particulier après toute modification de la dose de Lojuxta. La dose de warfarine doit être ajustée en cas d'indication clinique.
Consommation d'alcool
L'alcool peut augmenter les taux de graisses dans le foie et induire ou exacerber une atteinte hépatique. Dans l'étude de phase 3, 3 des 4 patients présentant des augmentations de l'ALAT au-delà de 5 fois la limite supérieure de la normale ont déclaré consommer de l'alcool en quantité supérieure aux limites recommandées dans le protocole. Il n'est pas recommandé de consommer de l'alcool pendant le traitement par lomitapide.
Agents hépatotoxiques
Il convient d'être prudent lorsque Lojuxta est utilisé avec d'autres médicaments connus pour être potentiellement hépatotoxiques, comme l'isotrétinoïne, l'amiodarone, le paracétamol (plus de 4 g/jour pendant 3 jours ou plus par semaine), le méthotrexate, les tétracyclines et le tamoxifène. L'effet de l'administration concomitante de lomitapide et d'autres médicaments hépatotoxiques n'est pas connu. Une surveillance plus fréquente de la fonction hépatique est conseillée.
Absorption réduite des vitamines liposolubles et des acides gras sériques
Étant donné son mode d'action dans l'intestin grêle, le lomitapide peut réduire l'absorption des nutriments liposolubles. Dans l'étude de phase 3, les patients ont reçu quotidiennement des compléments alimentaires de vitamine E, d'acide linoléique, d'AAL, d'AEP et d'ADH. Dans cette étude, les taux sériques médians de vitamine E, d'AAL, d'acide linoléique, d'AEP, d'ADH, et d'acide arachidonique avaient baissé par rapport aux valeurs avant traitement jusqu'à la semaine 26, mais se sont maintenus au-dessus de la limite inférieure de l'intervalle de référence. Il n'a pas été observé de conséquences cliniques indésirables de ces réductions pour une durée de traitement allant jusqu'à 78 semaines. Les patients sous Lojuxta doivent prendre quotidiennement des compléments alimentaires contenant 400 unités internationales de vitamine E et approximativement 200 mg d'acide linoléique, 210 mg d'AAL, 110 mg d'AEP et 80 mg d'ADH.
Mesures de contraception chez les femmes en âge de procréer
Avant d'instaurer le traitement chez les femmes en âge de procréer, des conseils appropriés concernant les méthodes efficaces de contraception doivent être donnés et une contraception efficace doit être prescrite. Les patientes qui prennent des contraceptifs oraux à base d'œstrogènes doivent être averties d'une éventuelle perte d'efficacité en raison de diarrhées et/ou de vomissements (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). Les contraceptifs oraux à base d'œstrogènes sont des inhibiteurs faibles du CYP3A4 (voir rubrique Posologie et mode d'administration).
En cas de grossesse, les patientes doivent immédiatement contacter leur médecin et arrêter le traitement (voir rubrique Fertilité, grossesse et allaitement).
Excipients à effet notoire
Lactose
Lojuxta contient du lactose. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par gélule, c.-à-d. qu'il est essentiellement « sans sodium ».
Résumé du profil de sécurité
Les effets indésirables les plus graves pendant le traitement étaient des taux anormaux des aminotransférases (voir rubrique Mises en garde spéciales et précautions d'emploi).
Les réactions indésirables les plus fréquentes étaient des effets gastro-intestinaux, rapportés chez 27 (93 %) des 29 patients dans l'étude clinique de phase 3. Des diarrhées sont survenues chez 79 % des patients, des nausées chez 65 %, une dyspepsie chez 38 % et des vomissements chez 34 %. D'autres effets rapportés chez au moins 20 % des patients comprennent des douleurs abdominales, une gêne abdominale, une distension abdominale, de la constipation et de la flatulence. Les effets indésirables gastro-intestinaux sont survenus plus fréquemment pendant la phase d'augmentation de doses par paliers de l'étude et diminuaient une fois que la dose maximale de lomitapide tolérée était déterminée chez les patients.
Des effets indésirables gastro-intestinaux d'intensité sévère ont été signalés par 6 (21 %) des 29 patients dans l'étude clinique de phase 3, les plus fréquents étant des diarrhées (4 patients, 14 %), des vomissements (3 patients, 10 %), ainsi que des douleurs, une distension et/ou une gêne abdominales (2 patients, 7 %). Des effets gastro-intestinaux faisaient partie des motifs de sortie prématurée de l'étude pour 4 patients (14 %).
Les effets indésirables d'intensité sévère les plus fréquemment rapportés étaient des diarrhées (4 sujets, 14 %), des vomissements (3 patients, 10 %), ainsi qu'une distension abdominale et une augmentation des taux d'ALAT (pour chaque effet, 2 sujets, 7 %).
Liste des effets indésirables sous forme de tableau
Les effets indésirables sont listés ci-dessous par CSO (Classe de Systèmes d'Organes) et par fréquence, à commencer par les plus fréquents. La fréquence des effets indésirables est définie comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Le Tableau 3 contient la liste de tous les effets indésirables rapportés pour l'ensemble des 35 patients traités dans l'étude de phase 2 UP1001 et dans l'étude de phase 3 UP1002/AEGR-733-005 ou son étude d'extension AEGR-733-012.
Tableau 3 : Fréquence des effets indésirables chez les patients présentant une HFHo
|
Classe de Systèmes d'Organes |
Fréquence |
Effet indésirable |
|
Infections et infestations |
Fréquent |
Gastro-entérite |
|
Troubles du métabolisme et de la nutrition |
Très fréquent |
Diminution de l'appétit |
|
Fréquence indéterminée |
Déshydratation |
|
|
Affections du système nerveux |
Fréquent |
Sensations vertigineuses Maux de tête Migraine |
|
Affections gastro-intestinales |
Très fréquent |
Diarrhées Nausées Vomissements Gêne abdominale Dyspepsie Douleurs abdominales Douleurs abdominales supérieures Flatulence Distension abdominale Constipation |
|
Fréquent |
Gastrite Ténesme rectal Aérophagie Besoin urgent d'aller à la selle Éructation Selles fréquentes Dilatation gastrique Trouble gastrique Reflux gastro-œsophagien Hémorragie hémorroïdaire Régurgitation |
|
|
Affections hépatobiliaires |
Fréquent |
Stéatose hépatique Hépatotoxicité Hépatomégalie |
|
Affections de la peau et du tissu sous-cutané |
Fréquent |
Ecchymoses Papule Éruption érythémateuse Xanthome |
|
Fréquence indéterminée |
Alopécie |
|
|
Affections musculo-squelettiques et du tissu conjonctif |
Fréquence indéterminée |
Myalgie |
|
Troubles généraux et anomalies au site d'administration |
Fréquent |
Fatigue |
|
Classe de Systèmes d'Organes |
Fréquence |
Effet indésirable |
|
Investigations |
Très fréquent |
Augmentation de l'alanine aminotransférase Augmentation de l'aspartate aminotransférase Perte de poids |
|
Fréquent |
Augmentation du rapport international normalisé Augmentation des phosphatases alcalines sanguines Diminution du potassium sanguin Diminution du carotène Rapport international normalisé anormal Tests de la fonction hépatique anormaux Allongement du temps de prothrombine Augmentation des transaminases Diminution de la vitamine E Diminution de la vitamine K |
Le tableau 4 énumère tous les effets indésirables pour les sujets ayant reçu du lomitapide en monothérapie (N = 291) dans les études de phase 2 menées chez des sujets présentant des taux élevés de C-LDL (N = 462).
Tableau 4 : Fréquence des effets indésirables chez les patients présentant des taux élevés de C-LDL
|
Classe de Systèmes d'Organes |
Fréquence |
Effet indésirable |
|
Infections et infestations |
Peu fréquent |
Gastro-entérite Infection gastro-intestinale Grippe Rhinopharyngite Sinusite |
|
Affections hématologiques et du système lymphatique |
Peu fréquent |
Anémie |
|
Troubles du métabolisme et de la nutrition |
Fréquent |
Diminution de l'appétit |
|
Peu fréquent |
Déshydratation Augmentation de l'appétit |
|
|
Affections du système nerveux |
Peu fréquent |
Paresthésie Somnolence |
|
Affections oculaires |
Peu fréquent |
Gonflement des yeux |
|
Affections de l'oreille et du labyrinthe |
Peu fréquent |
Vertiges |
|
Affections respiratoires, thoraciques et médiastinales |
Peu fréquent |
Lésion pharyngée Syndrome de toux des voies aériennes supérieures |
|
Classe de Systèmes d'Organes |
Fréquence |
Effet indésirable |
|
Affections gastro-intestinales |
Très fréquent |
Diarrhées Nausées Flatulence |
|
Fréquent |
Douleurs abdominales supérieures Distension abdominale Douleurs abdominales Vomissements Gêne abdominale Dyspepsie Éructation Douleurs abdominales inférieures Selles fréquentes |
|
|
Peu fréquent |
Sécheresse buccale Selles dures Reflux gastro-œsophagien Sensibilité abdominale Gêne épigastrique Dilatation gastrique Hématémèse Hémorragie gastro-intestinale inférieure Œsophagite par reflux gastro-œsophagien |
|
|
Affections hépatobiliaires |
Peu fréquent |
Hépatomégalie |
|
Affections de la peau et du tissu sous-cutané |
Peu fréquent |
Ampoule Peau sèche Hyperhidrose |
|
Affections musculo-squelettiques et du tissu conjonctif |
Fréquent |
Spasmes musculaires |
|
Peu fréquent |
Arthralgie Myalgie Douleurs des extrémités Gonflement des articulations Secousse musculaire |
|
|
Affections du rein et des voies urinaires |
Peu fréquent |
Hématurie |
|
Troubles généraux et anomalies au site d'administration |
Fréquent |
Fatigue Asthénie |
|
Peu fréquent |
Douleur dans la poitrine Frissons Sensation de satiété précoce Troubles de la démarche Malaise Pyrexie |
|
|
Classe de Systèmes d'Organes |
Fréquence |
Effet indésirable |
|
Investigations |
Fréquent |
Augmentation de l'alanine aminotransférase Augmentation de l'aspartate aminotransférase Augmentation des enzymes hépatiques Tests de la fonction hépatique anormaux Diminution du nombre de neutrophiles Diminution du nombre de globules blancs |
|
Peu fréquent |
Perte de poids Augmentation de la bilirubine sanguine Augmentation de la gammaglutamyltransférase Augmentation du pourcentage de neutrophiles Protéinurie Allongement du temps de prothrombine Test de la fonction pulmonaire anormal Augmentation du nombre de globules blancs |
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé doivent déclarer tout effet indésirable suspecté via le système national de déclaration - voirAnnexe V.
AVANT la mise en route du traitement :
- CONFIRMER le diagnostic d'une hypercholestérolémie familiale homozygote par un test génétique dans toute la mesure du possible.
- CONFIRMER l'absence de grossesse chez les femmes en âge de procréer.
SURVEILLANCE du traitement :
- Phosphatases alcalines, bilirubine totale, gamma-glutamyl transférase (gamma-GT), l'albumine sérique.
- ASAT/ALAT : pendant la première année de traitement, avant chaque augmentation ou au moins une fois par mois. Après la première année de traitement, au moins tous les 3 mois et avant chaque augmentation de dose.
- Dépistage d'une stéato-hépatite ou d'une fibrose avant le traitement puis annuellement.
ADMINISTRER quotidiennement, pendant toute la durée du traitement, des compléments alimentaires apportant :
- 400 UI de vitamine E et approximativement 200 mg d'acide linoléique,
- 110 mg d'acide eicosapentaénoïque (AEP),
- 210 mg d'acide alpha linolénique (AAL),
- 80 mg d'acide docosahexaénoïque (ADH).
PATIENTES EN AGE DE PROCREER : Prescrire une méthode efficace de contraception.
- CONFIRMER le diagnostic d'une hypercholestérolémie familiale homozygote par un test génétique dans toute la mesure du possible.
- CONFIRMER l'absence de grossesse chez les femmes en âge de procréer.
SURVEILLANCE du traitement :
- Phosphatases alcalines, bilirubine totale, gamma-glutamyl transférase (gamma-GT), l'albumine sérique.
- ASAT/ALAT : pendant la première année de traitement, avant chaque augmentation ou au moins une fois par mois. Après la première année de traitement, au moins tous les 3 mois et avant chaque augmentation de dose.
- Dépistage d'une stéato-hépatite ou d'une fibrose avant le traitement puis annuellement.
ADMINISTRER quotidiennement, pendant toute la durée du traitement, des compléments alimentaires apportant :
- 400 UI de vitamine E et approximativement 200 mg d'acide linoléique,
- 110 mg d'acide eicosapentaénoïque (AEP),
- 210 mg d'acide alpha linolénique (AAL),
- 80 mg d'acide docosahexaénoïque (ADH).
PATIENTES EN AGE DE PROCREER : Prescrire une méthode efficace de contraception.
Lors de l'utilisation de pilules contraceptives
et en cas d'épisode de diarrhées, ou de vomissements qui durent plus de 2
jours, UTILISER une autre méthode de contraception (p. ex. des préservatifs, un
diaphragme) pendant 7 jours après la disparition des symptômes.
INFORMER IMMEDIATEMENT LE MEDECIN en cas de nausée, vomissement, douleur à l'estomac, douleurs musculaires, fièvre, jaunissement de la peau ou du blanc des yeux, fatigue plus intense que d'habitude, sensation d'avoir la grippe.
NE PAS consommer de jus de pamplemousse pendant le traitement.
EVITER de consommer du millepertuis (Hypericum perforatum) pendant le traitement.
EVITER la consommation d'alcool pendant le traitement
PREVENIR LE MEDECIN en cas de consommation d'huile de menthe poivrée ou d'oranges amères ; le traitement pourra être ajusté.
INFORMER IMMEDIATEMENT LE MEDECIN en cas de nausée, vomissement, douleur à l'estomac, douleurs musculaires, fièvre, jaunissement de la peau ou du blanc des yeux, fatigue plus intense que d'habitude, sensation d'avoir la grippe.
NE PAS consommer de jus de pamplemousse pendant le traitement.
EVITER de consommer du millepertuis (Hypericum perforatum) pendant le traitement.
EVITER la consommation d'alcool pendant le traitement
PREVENIR LE MEDECIN en cas de consommation d'huile de menthe poivrée ou d'oranges amères ; le traitement pourra être ajusté.
Utilisation chez les femmes en âge de procréer
Avant d'instaurer le traitement chez les femmes en âge de procréer, l'absence de grossesse doit être confirmée, des conseils appropriés relatifs aux méthodes de contraception efficaces doivent être dispensés et une contraception efficace doit être prescrite. Les patientes prenant des contraceptifs oraux à base d'œstrogènes doivent être averties d'une diminution possible de leur efficacité en raison de diarrhées et/ou de vomissements. Des mesures contraceptives supplémentaires doivent être prises, jusqu'à la disparition des symptômes (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Grossesse
Lojuxta est contre-indiqué pendant la grossesse. Il n'existe pas de données fiables concernant son utilisation chez les femmes enceintes. Des études animales ont montré une toxicité sur le développement (tératogénicité, embryotoxicité, voir rubrique Données de sécurité préclinique). Le risque potentiel chez l'homme n'est pas connu.
Allaitement
On ne sait pas si le lomitapide est excrété dans le lait maternel. En raison d'effets indésirables possibles au vu de résultats obtenus dans des études menées avec le lomitapide chez l'animal (voir rubrique Données de sécurité préclinique), il convient de prendre la décision soit d'interrompre l'allaitement soit d'arrêter le médicament, en tenant compte de l'importance du médicament pour la mère.
Fertilité
Aucun effet indésirable sur la fertilité n'a été observé chez des rats mâles et femelles ayant reçu des doses de lomitapide conduisant à des expositions systémiques (ASC) estimées 4 à 5 fois plus intenses que celle produite chez l'homme par la dose maximale recommandée (voir rubrique Données de sécurité préclinique).
Effets d'autres médicaments sur le lomitapide et autres formes d'interactions
Tableau 2 : Interactions entre Lojuxta et d'autres médicaments et autres formes d'interactions
|
Médicaments |
Effets sur les taux de lomitapide |
Recommandation concernant l'administration concomitante avec Lojuxta |
|
Inhibiteurs du CYP3A4 |
Inhibiteurs puissants et modérés Lorsque 60 mg de lomitapide ont été administrés avec 200 mg, deux fois par jour, de kétoconazole, un inhibiteur puissant du CYP3A4, l'ASC du lomitapide a augmenté d'un facteur 27 environ et la Cmax augmenté de près de 15 fois. Les interactions entre des inhibiteurs modérés du CYP3A4 et le lomitapide n'ont pas été étudiées. Les inhibiteurs modérés du CYP3A4 pourraient avoir un effet important sur les propriétés pharmacocinétiques du lomitapide. Une utilisation concomitante d'inhibiteurs modérés du CYP3A4 est susceptible d'augmenter l'exposition au lomitapide d'un facteur 4 à 10, au vu des résultats de l'étude menée avec le kétoconazole, un inhibiteur puissant du CYP3A4, ainsi que des données historiques obtenues avec le modèle clinique utilisant le midazolam comme substrat du CYP3A4. |
Inhibiteurs puissants et modérés L'utilisation d'inhibiteurs puissants ou modérés du CYP3A4 avec Lojuxta est contre-indiquée. Si un traitement par des antifongiques azolés (p. ex. itraconazole, kétoconazole, fluconazole, voriconazole, posaconazole), l'antiarythmique dronédarone, des antibiotiques macrolides (p. ex. érythromycine, clarithromycine), des antibiotiques kétolides (par ex. télithromycine), des inhibiteurs des protéases du VIH, les bloqueurs des canaux calciques diltiazem et vérapamil, ne peut être évité, Lojuxta doit être interrompu pendant la durée du traitement (voir rubriques Contre-indications et Mises en garde spéciales et précautions d'emploi). Le jus de pamplemousse est un inhibiteur modéré du CYP3A4 et pourrait augmenter de façon importante l'exposition au lomitapide. Les patients sous Lojuxta doivent éviter de consommer du jus de pamplemousse. |
|
|
Inhibiteurs faibles Les inhibiteurs faibles du CYP3A4 sont susceptibles d'augmenter l'exposition au lomitapide lorsqu'ils sont pris de façon concomitante. Lors de l'administration concomitante de 20 mg de lomitapide avec l'atorvastatine, un inhibiteur faible du CYP3A4, l'ASC et la Cmax du lomitapide ont augmenté approximativement d'un facteur 2. Lorsque la dose de lomitapide a été prise à 12 heures d'intervalle de l'atorvastatine, il n'a pas été observé d'augmentation cliniquement significative de l'exposition au lomitapide. Lors de l'administration concomitante, ou à 12 heures d'intervalle, de 20 mg de lomitapide avec l'éthinyl estradiol/norgestimate, un inhibiteur faible du CYP3A4, il n'a pas été observé d'augmentation cliniquement significative de l'exposition au lomitapide. |
Inhibiteurs faibles En cas d'administration avec l'atorvastatine, la dose de Lojuxta doit soit être prise à 12 heures d'intervalle soit être diminuée de moitié (voir rubrique Posologie et mode d'administration). La dose de Lojuxta doit être prise à 12 heures d'intervalle de n'importe quel autre inhibiteur faible du CYP3A4. Parmi les exemples d'inhibiteurs faibles du CYP3A4 figurent les produits suivants : alprazolam, amiodarone, amlodipine, atorvastatine, azithromycine, bicalutamide, cilostazol, cimétidine, ciclosporine, clotrimazole, fluoxétine, fluvoxamine, fosaprépitant, ginkgo, hydraste du Canada, isoniazide, ivacaftor, lacidipine, lapatinib, linagliptine, nilotinib, contraceptifs oraux à base d'œstrogènes, pazopanib, huile de menthe poivrée, propivérine, ranitidine, ranolazine, roxithromycine, orange amère, tacrolimus, ticagrélor et tolvaptan. Cette liste n'est pas exhaustive et les prescripteurs doivent vérifier les informations de prescription des médicaments devant être co-administrés avec Lojuxta, pour identifier des interactions potentielles dans lesquelles le CYP3A4 sert de médiateur. L'effet de l'administration de plus d'un inhibiteur faible du CYP3A4 n'a pas été testé, mais il est attendu que l'effet sur l'exposition au lomitapide soit plus forte que pour l'administration concomitante d'un seul inhibiteur avec le lomitapide. L'administration de Lojuxta avec plus d'un inhibiteur faible du CYP3A4 impose une prudence supplémentaire. |
|
Inducteurs du CYP3A4 |
Les médicaments inducteurs du CYP3A4 peuvent augmenter la vitesse et l'intensité du métabolisme du lomitapide. Par conséquent, l'effet du lomitapide serait réduit. Tout effet sur l'efficacité risque d'être variable. |
Lors de la co-administration d'inducteurs du CYP3A4 (p. ex. aminoglutéthimide, nafcilline, inhibiteurs non nucléosidiques de la transcriptase inverse, phénobarbital, rifampicine, carbamazépine, pioglitazone, millepertuis, glucocorticoïdes, modafinil et phénytoïne) et de Lojuxta, la possibilité d'une interaction médicamenteuse modifiant l'efficacité doit être prise en compte. Il est recommandé d'augmenter la fréquence des mesures du C-LDL pendant une telle utilisation concomitante et d'envisager d'augmenter la dose de Lojuxta pour assurer le maintien du niveau d'efficacité souhaité, s'il est prévu une utilisation chronique de l'inducteur du CYP3A4. |
|
Chélateurs des acides biliaires |
Le lomitapide n'a pas fait l'objet d'une étude d'interaction avec les chélateurs des acides biliaires (résines telles que le colésévélam et la cholestyramine). |
Les chélateurs des acides biliaires pouvant interférer avec l'absorption de médicaments oraux, ils doivent être pris au moins 4 heures avant ou au moins 4 h après Lojuxta. |
Effets du lomitapide sur d'autres médicaments
Inhibiteurs de l'HMG-CoA réductase (« statines »)
Le lomitapide augmente les concentrations plasmatiques des statines. L'administration de 40 mg de simvastatine après administration de 60 mg de lomitapide jusqu'à l'état d'équilibre montre une augmentation de l'ASC et de la Cmax de la simvastatine acide respectivement de 68 % et de 57 %. L'admistration de 20 mg d'atorvastatine après administration de 60 mg de lomitapide jusqu'à l'état d'équilibre montre une augmentation de l'ASC et de la Cmax de l'atorvastatine acide respectivement de 52 % et de 63 %.
L'administration de 20 mg de rosuvastatine après administration de 60 mg de lomitapide jusqu'à l'état d'équilibre montre une augmentation du Tmax de 1 à 4 heures, de l'ASC de 32 % et la Cmax est restée inchangée.
Le risque de myopathie avec la simvastatine est dose-dépendant. L'utilisation de Lojuxta est contre-indiquée chez les patients traités par de fortes doses de simvastatine (> à 40 mg) (voir rubriques Contre-indications et Mises en garde spéciales et précautions d'emploi).
Anticoagulants coumariniques
Lors de l'administration de 60 mg de lomitapide jusqu'à l'état d'équilibre, puis de 10 mg de warfarine pendant 6 jours, l'INR a augmenté d'un facteur 1,26. Les ASC pour la warfarine R(+) et la warfarine S(-) ont augmenté respectivement de 25 % et de 30 %. Les Cmax pour la warfarine R(+) et la warfarine S(-) ont augmenté respectivement de 14 % et de 15 %. Chez les patients prenant de façon concomitante des coumarines (tels que la warfarine) et Lojuxta, l'INR doit être déterminé avant de commencer le traitement par Lojuxta et surveillé régulièrement, la posologie des coumarines devant être ajustée en cas d'indication clinique (voir rubrique Mises en garde spéciales et précautions d'emploi).
Fénofibrate, niacine et ézétimibe
Lors de l'administration de 145 mg de fénofibrate micronisé, de 1 000 mg de niacine à libération prolongée ou de 10 mg d'ézétimibe après administration de lomitapide jusqu'à l'état d'équilibre, aucun effet cliniquement significatif sur l'exposition de ces médicaments n'a été observé. Il n'est pas nécessaire d'ajuster leurs doses lorsqu'ils sont administrés de façon concomitante avec Lojuxta.
Contraceptifs oraux
Lors de l'administration d'un contraceptif oral à base d'oestrogènes après administration de 50 mg de lomitapide jusqu'à l'état d'équilibre, il n'a été observé aucun effet cliniquement important ni statitisquement significatif sur les propriétés pharmacocinétiques des composants du contraceptif oral (éthinylestradiol et 17-désacétyl-norgestimate, le métabolite du norgestimate). Le lomitapide n'influe en principe pas directement sur l'efficacité des contraceptifs oraux à base d'œstrogènes ; cependant, des diarrhées et/ou des vomissements peuvent réduire l'absorption des hormones. En cas de diarrhées prolongées ou sévères et/ou de vomissements qui durent plus de 2 jours, des mesures contraceptives supplémentaires doivent être prises pendant 7 jours après la disparition des symptômes.
Substrats de la P-gp
Le lomitapide inhibe la P-gp in vitro et peut augmenter l'absorption des substrats de la P-gp. L'administration concomitante de Lojuxta et de substrats de la P-gp (tels que les médicaments suivants : aliskiren, ambrisentan, colchicine, dabigatran étexilate, digoxine, évérolimus, fexofénadine, imatinib, lapatinib, maraviroc, nilotinib, posaconazole, ranolazine, saxagliptine, sirolimus, sitagliptine, talinolol, tolvaptan, topotécan) peut augmenter l'absorption des substrats de la P-gp. Il convient d'envisager une réduction de la dose du substrat de la P-gp lors d'une utilisation concomitante avec Lojuxta.
Évaluation in vitro d'interactions médicamenteuses
Le lomitapide inhibe le CYP3A4. Il n'induit pas les CYP 1A2, 3A4 ou 2B6 et n'inhibe pas les CYP 1A2, 2B6, 2C9, 2C19, 2D6 ou 2E1. Il n'est pas un substrat de la P-gp, mais il inhibe la P-gp. Par contre, il n'inhibe pas la protéine de résistance au cancer du sein (BCRP, breast cancer resistance protein).
Le traitement par Lojuxta doit être instauré et surveillé par un médecin qualifié dans la prise en charge des patients atteints de dyslipidémie.
Posologie
La posologie initiale recommandée est de 5 mg une fois par jour. Après 2 semaines, la dose peut être augmentée, en fonction de la réponse en C-LDL et sur la base d'une sécurité et d'une tolérance acceptables, à 10 mg, puis, à des intervalles d'au moins 4 semaines, à 20 mg, 40 mg et jusqu'à la dose maximale recommandée de 60 mg (voir rubrique Mises en garde spéciales et précautions d'emploi).
La dose doit être augmentée progressivement, afin de minimiser l'incidence et la gravité des effets indésirables gastro-intestinales et l'augmentation des aminotransférases.
La survenue et la gravité des effets indésirables gastro-intestinaux associés à Lojuxta diminuent avec un régime alimentaire pauvre en graisses. Les patients doivent suivre un régime apportant moins de 20 % des calories sous forme de graisses avant de commencer le traitement et poursuivre ce régime pendant le traitement. Les patients doivent recevoir des conseils diététiques.
Les patients doivent éviter de consommer du jus de pamplemousse et de l'alcool (voir rubriques Mises en garde spéciales et précautions d'emploi et Interactions avec d'autres médicaments et autres formes d'interactions).
Chez les patients traités par une dose d'entretien stable de Lojuxta qui reçoivent de l'atorvastatine, il faut soit :
- Espacer la prise des médicaments de 12 heures
OU
- Réduire la dose de Lojuxta de moitié. Les patients traités par 5 mg doivent rester à 5 mg.
Une augmentation de la dose peut ensuite être envisagée avec prudence en fonction de la réponse en C-LDL et de la sécurité/tolérance. Lors de l'arrêt de l'atorvastatine, la dose de Lojuxta doit être augmentée progressivement par paliers, en fonction de la réponse en C-LDL et de la sécurité/tolérance.
Chez les patients traités par une dose d'entretien stable de Lojuxta, qui reçoivent n'importe quel autre inhibiteur faible du cytochrome P450 (CYP) 3A4, il faut espacer la prise des médicaments (Lojuxta et l'inhibiteur faible du CYP3A4) de 12 heures. Des précautions supplémentaires s'imposent lors de l'administration de Lojuxta avec plus d'un inhibiteur faible du CYP3A4. La dose maximale de Lojuxta doit être limitée en fonction de la réponse en C-LDL souhaitée.
Une diminution des taux d'acides gras essentiels et de vitamine E dans les études cliniques a été observée, les patients doivent prendre quotidiennement des compléments alimentaires apportant 400 UI de vitamine E et approximativement 200 mg d'acide linoléique, 110 mg d'acide eicosapentaénoïque (AEP), 210 mg d'acide alpha linolénique (AAL) et 80 mg d'acide docosahexaénoïque (ADH), pendant toute la durée du traitement par Lojuxta (voir rubrique Mises en garde spéciales et précautions d'emploi).
Populations particulières
Patients âgés
L'expérience du traitement par lomitapide chez les patients âgés de 65 ans ou plus est limitée. Il convient donc d'être particulièrement prudent chez ces patients.
La posologie recommandée implique de débuter le traitement à la dose la plus faible et d'augmenter prudemment en fonction de la tolérance individuelle des patients, ainsi aucun ajustement de la posologie n'est recommandé chez les personnes âgées.
Insuffisance hépatique
Le lomitapide est contre-indiqué chez les patients présentant une insuffisance hépatique modérée ou sévère, y compris les patients dont les tests de la fonction hépatique sont anormaux de façon prolongée et inexpliquée (voir rubriques Contre-indications et Propriétés pharmacocinétiques).
Les patients souffrant d'une insuffisance hépatique légère (Child-Pugh A) ne doivent pas dépasser 40 mg par jour.
Insuffisance rénale
Les patients présentant une maladie rénale en phase terminale, traités par dialyse, ne doivent pas dépasser 40 mg par jour (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l'efficacité du lomitapide n'ont pas été établies chez les enfants âgés de moins de 18 ans et l'utilisation de ce médicament chez les enfants n'est donc pas recommandée. Aucune donnée n'est disponible.
Mode d'administration
Voie orale.
L'administration avec des aliments peut augmenter l'exposition au lomitapide. Il doit donc être pris à jeun, au moins 2 heures après le repas du soir, car la teneur en graisses d'un repas récent peut avoir un effet défavorable sur tolérance gastro-intestinale (voir rubrique Mises en garde spéciales et précautions d'emploi).
Durée de conservation :
3 ans.
Précautions particulières de conservation :
À conserver à une température ne dépassant pas 30 °C.
Conserver le flacon soigneusement fermé à l'abri de l'humidité.
Sans objet.
Il n'existe pas d'antidote spécifique en cas de surdosage. En cas de surdosage, traiter les symptômes du patient et mettre en place des mesures de soutien selon les besoins. Surveiller les analyses des taux hépatiques. Une hémodialyse n'est pas susceptible d'être bénéfique puisque le lomitapide se lie fortement aux protéines.
Chez les rongeurs, des doses orales uniques de lomitapide ≥ à 600 fois la dose maximale recommandée chez l'homme (1 mg/kg) étaient bien tolérées. La dose maximale administrée à l'homme dans les études cliniques était de 200 mg en dose unique ; il n'y a pas eu d'effets indésirables.
Classe pharmacothérapeutique : agents modifiant les lipides, autres agents modifiant les lipides. Code ATC : C10AX12
Mécanisme d'action
Le lomitapide est un inhibiteur sélectif de la protéine microsomale de transfert des triglycérides (PMT), une protéine intracellulaire de transfert de lipides présente dans le lumen du réticulum endoplasmique et qui assure la liaison et le transport de molécules individuelles de lipides entre les membranes. La PMT joue un rôle clé dans l'assemblage des lipoprotéines contenant de l'Apo B dans le foie et les intestins. L'inhibition de la PMT diminue la sécrétion de lipoprotéines et les concentrations circulantes de lipides transportés par des lipoprotéines, notamment du cholestérol et des triglycérides.
Efficacité et sécurité clinique
Une étude en ouvert à un seul bras (UP1002/AEGR-733-005) visait à évaluer l'efficacité et la sécurité du lomitapide, co-administré avec un régime alimentaire pauvre en graisses et d'autres traitements hypolipémiants, chez des patients adultes présentant une HFHo. Les patients devaient maintenir un régime pauvre en graisses (apport de moins de 20 % des calories par les graisses) et leurs traitements hypolipémiants à l'entrée dans l'étude, y compris une aphérèse le cas échéant, à partir de 6 semaines avant la détermination de la valeur de base et au moins jusqu'à la semaine 26. La dose de lomitapide a été augmentée progressivement, de 5 mg jusqu'à une dose maximale tolérée déterminée pour chaque patient individuellement et jusqu'à un maximum de 60 mg. Après la semaine 26, les patients ont été maintenus sous lomitapide, afin de déterminer les effets du traitement à long terme et étaient autorisés à changer de traitements hypolipémiants de fond. La durée totale de l'étude prévue a été de 78 semaines.
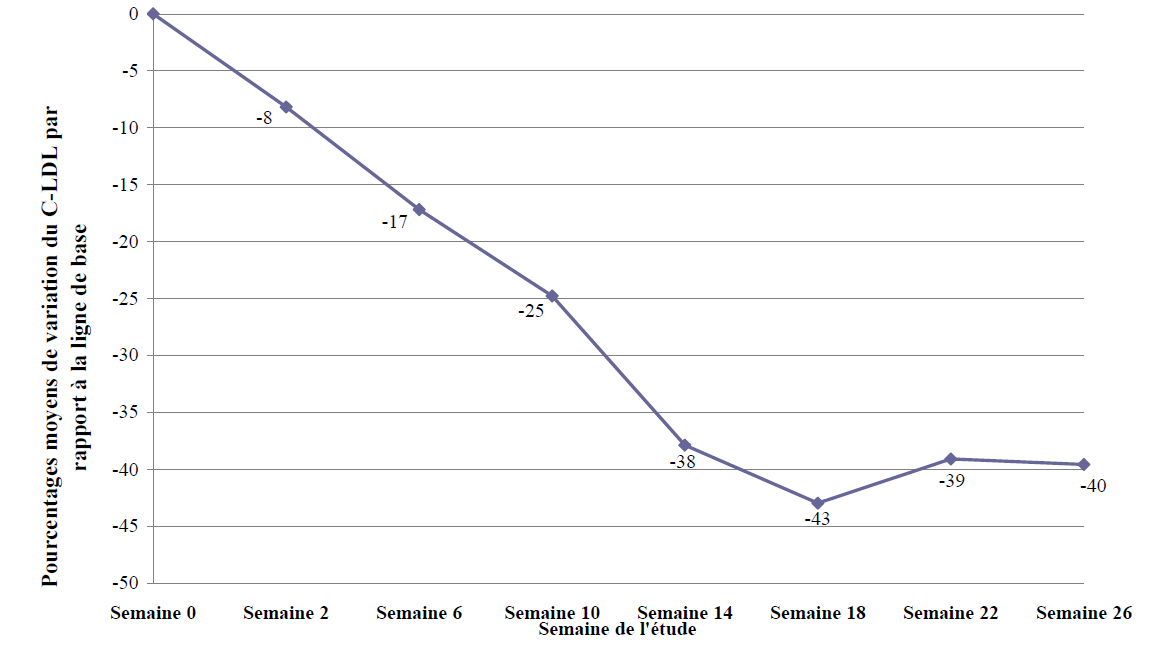
Vingt-neuf patients ont été inclus, parmi lesquels 23 ont terminé l'étude à 78 semaines. Seize hommes (55 %) et 13 femmes (45 %) ont été inclus, d'un âge moyen de 30,7 ans, pour un intervalle de 18 à 55 ans. La dose moyenne de lomitapide était de 45 mg à la semaine 26 et de 40 mg à la semaine 78. À la semaine 26, le pourcentage moyen de variation du C-LDL par rapport à la valeur de base était de - 40 % (p < 0,001) dans la population en intention de traitement (population ITT). Le pourcentage moyen de variation par rapport à la valeur de base jusqu'à la semaine 26, calculé à l'aide des dernières observations rapportées (LOCF, last observation carried forward) pour chaque évaluation est représenté en figure 1.
Figure 1 : Pourcentages moyens de variation du C-LDL par rapport à la valeur de base dans l'étude principale d'efficacité UP1002/AEGR-733-005 jusqu'à la semaine 26 (critère principal d'évaluation), calculés à l'aide des LOCF pour chaque évaluation (N = 29)
Les variations des concentrations en lipides et en lipoprotéines observées aux semaines 26 et 78 du traitement par le lomitapide sont présentées dans le Tableau 5.
Tableau 5 : Variations en valeurs absolues et en pourcentages par rapport à la valeur de base des concentrations en lipides et en lipoprotéines, observées aux semaines 26 et 78 (étude principale d'efficacité UP1002/AEGR-733-005)
|
Paramètre (unités) |
Ligne de base |
Semaine 26/LOCF (N = 29) |
Semaine 78 (N = 23) |
||||
|
Moyenne (ET) |
Moyenne (ET) |
% de variation |
Valeur de p b |
Moy enne (ET) |
% de variation |
Valeur de p b |
|
|
C-LDL, direct (mg/dl) |
336 (114) |
190 (104) |
-40 |
<0,001 |
210 (132) |
-38 |
<0,001 |
|
Cholestérol total (CT) (mg/dl) |
430 (135) |
258 (118) |
-36 |
<0,001 |
281 (149) |
-35 |
<0,001 |
|
Apolipoprotéine B (Apo B) (mg/dl) |
259 (80) |
148 (74) |
-39 |
<0,001 |
151 (89) |
-43 |
<0,001 |
|
Triglycérides (TG) (mg/dl)a |
92 |
57 |
-45 |
0,009 |
59 |
-42 |
0,012 |
|
Cholestérol des lipoprotéines autres que de haute densité (C non-HDL) (mg/dl) |
386 (132) |
217 (113) |
-40 |
<0,001 |
239 (146) |
-39 |
<0,001 |
|
Cholestérol des lipoprotéines de très basse densité (CVLDL) (mg/dl) |
21 (10) |
13 (9) |
-29 |
0,012 |
16 (15) |
-31 |
0,013 |
|
Lipoprotéine (a) (Lp(a)) (nmol/l)a |
66 |
61 |
-13 |
0,094 |
72 |
-4 |
<0,842 |
|
Cholestérol des lipoprotéines de haute densité (C-HDL) (mg/dl) |
44 (11) |
41 (13) |
-7 |
0,072 |
43 (12) |
-4,6 |
0,246 |
a Médiane présentée pour les TG et la Lp(a). La valeur de p est basée sur le pourcentage moyen des variations.
b Valeur de p basée sur le pourcentage moyen des variations par rapport à la valeur de base pour le test t pour échantillons appariés.
À la semaine 26 ainsi qu'à la semaine 78, des diminutions significatives ont été observées pour le C-LDL, le CT, l'Apo B, les TG, le C non-HDL le C-VLDL ; les variations du C-HDL montrent une tendance à la baisse à la semaine 26 et un retour aux niveaux des valeurs de base à la semaine 78.
L'effet de Lojuxta sur la morbidité et la mortalité cardiovasculaires n'a pas été déterminé.
Au début de l'étude, 93 % des patients prenaient une statine, 76 % étaient sous ézétimibe, 10 % sous niacine, 3 % prenaient un chélateur des acides biliaires et 62 % recevaient un traitement par aphérèse. Chez 15 des 23 patients (65 %), le traitement hypolipémiant a été réduit à la semaine 78 incluant réductions/interruptions/ prévues et non prévues. L'aphérèse a été arrêtée chez 3 des 13 patients qui en bénéficiaient à la semaine 26 et sa fréquence a été réduite chez 3 patients, chez lesquels les taux de C-LDL sont restés bas jusqu'à la semaine 78. Le bénéfice clinique des réductions du traitement hypolipémiant de fond, y compris de l'aphérèse, n'est pas certain.
Parmi les 23 patients restés dans l'étude jusqu'à la semaine 78, 19 (83 %) présentaient des baisses du C-LDL ≥ à 25 %, avec 8 patients (35 %) présentant des taux de C-LDL < à 100 mg/dl et 1 patient ayant un C-LDL < à 70 mg/dl à ce stade de l'étude.
Dans cette étude, 10 patients ont présenté des augmentations de l'ASAT et/ou de l'ALAT > à 3 fois la LSN (voir Tableau 6).
Tableau 6 : Résultats les plus élevés observés pour les tests de la fonction hépatique après la première dose (étude principale d'efficacité UP1002/AEGR-733-005)
|
Paramètre/anomalie |
N (%) |
|
ALAT |
|
|
Nombre de patients évalués |
29 |
|
> à 3 et ≤ à 5 x LSN |
6 (20,7) |
|
> à 5 et ≤ à 10 x LSN |
3 (10,3) |
|
> à 10 et ≤ à 20 x LSN |
1 (3,4) |
|
> à 20 x LSN |
0 |
|
ASAT |
|
|
Nombre de patients évalués |
29 |
|
> à 3 et ≤ à 5 x LSN |
5 (17,2) |
|
> à 5 et ≤ à 10 x LSN |
1 (3,4) |
|
> à 10 et ≤ à 20 x LSN |
0 |
|
> à 20 x LSN |
0 |
Les augmentations de l'ALAT et/ou de l'ASAT > à 5 fois la LSN ont été prises en charge par une réduction de la dose ou une suspension temporaire de la prise de lomitapide et tous les patients ont pu poursuivre le traitement par le médicament à l'étude. Aucune augmentation cliniquement importante de la bilirubine totale ou des phosphatases alcalines n'a été observée. Les graisses hépatiques ont fait l'objet d'une mesure prospective par spectroscopie par RMN chez tous les patients éligibles au cours de l'étude clinique (Tableau 7). Les données des patients chez lesquelles les mesures ont été répétées après l'arrêt du lomitapide montrent que l'accumulation de graisses dans le foie est réversible, mais on ne sait pas s'il subsiste des séquelles histologiques.
Tableau 7 : Variations catégoriques maximales du pourcentage de graisses hépatique (étude principale d'efficacité UP1002/AEGR-733-005)
|
Augmentation absolue maximale des graisses hépatiques en % |
Phase efficacité semaines 0 à 26 N (%) |
Phase sécurité semaines 26 à 78 N (%) |
Essai complet semaines 0 à 78 N (%) |
|
Nombre de patients évaluables |
22 |
22 |
23 |
|
≤ à 5 % |
9 (41) |
6 (27) |
5 (22) |
|
> à 5 % et ≤ à 10 % |
6 (27) |
8 (36) |
8 (35) |
|
> à 10 % et ≤ à 15 % |
4 (18) |
3 (14) |
4 (17) |
|
> à 15 % et ≤ à 20 % |
1 (5) |
4 (18) |
3 (13) |
|
> à 20 % et ≤ à 25 % |
1 (5) |
0 |
1 (4) |
|
> à 25 % |
1 (5) |
1 (5) |
2 (9) |
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec Lojuxta dans un ou plusieurs sous-groupes de la population pédiatrique dans l'indication d'HFHo (voir rubrique Posologie et mode d'administration pour des informations concernant l'usage pédiatrique).
Une autorisation de mise sur le marché « sous circonstances exceptionnelles » a été délivrée pour ce médicament. Cela signifie qu'en raison de la rareté de cette maladie il n'a pas été possible d'obtenir des informations complètes concernant ce médicament.
L'Agence européenne du médicament réévaluera chaque année toute nouvelle information qui pourrait être disponible, et, si nécessaire, ce RCP sera mis à jour.
Absorption
La biodisponibilité orale absolue du lomitapide est de 7 %. L'absorption n'est pas limitée par le passage du principe actif à travers la barrière intestinale, mais elle est principalement influencée par un intense effet de premier passage. Les pics des concentrations plasmatiques de lomitapide étaient atteints 4 à 8 heures après la prise orale. Les propriétés pharmacocinétiques du lomitapide sont presque proportionnelles à la dose pour des doses orales uniques dans l'intervalle thérapeutique. Des doses supérieures à 60 mg suggèrent une tendance à la non linéarité et ne sont pas recommandées.
Après la prise de doses répétées, la Cmax et l'ASC ont augmenté dans une proportion proche de la dose de lomitapide. La Cmax et l'ASC augmentent à la suite d'un repas riche en graisses (respectivement de 77 % et 58 %) ou d'un repas pauvre en graisses (respectivement de 70 % et 28 %). L'accumulation de lomitapide dans le plasma était conforme à celle prévue après administration par voie orale d'une dose unique, supérieure à 25 mg pendant 4 semaines. La variabilité interindividuelle de l'ASC du lomitapide était de 50 % environ.
À l'état d'équilibre, l'accumulation du lomitapide était de 2,7 pour 25 mg et de 3,9 pour 50 mg.
Distribution
Après une administration intraveineuse, le volume de distribution du lomitapide était élevé (moyenne = 1 200 litres), malgré un fort degré (> 99,8 %) de liaison aux protéines plasmatiques. Dans les études chez l'animal, le lomitapide était fortement concentré (200 fois) dans le foie.
Biotransformation
Le lomitapide est considérablement métabolisé, principalement par le CYP3A4. Les isoformes de CYP 2E1, 1A2, 2B6, 2C8 et 2C19 interviennent dans une moindre mesure et les isoformes 2D6 et 2C9 ne participent pas au métabolisme du lomitapide.
Élimination
Après l'administration d'une solution orale radiomarquée à des sujets sains, 93 % de la dose administrée étaient retrouvés dans les urines et les fèces. Environ 33 % de la radioactivité était excrétée dans les urines sous forme de métabolites. Le reste était excrété dans les fèces, principalement sous forme de métabolites oxydés. La demi-vie d'élimination du lomitapide était de 29 heures environ.
Populations spéciales
Les données de l'étude clinique pivot ont été analysées en tenant compte de l'impact de covariables potentielles sur l'exposition au lomitapide. Parmi les paramètres étudiés (origine ethnique, indice de masse corporelle (IMC), sexe, poids, âge), seul l'IMC a pu être classé comme covariable potentielle.
Âge et sexe
Il n'y avait pas d'effet cliniquement significatif de l'âge (18 à 64 ans) ni du sexe sur les propriétés pharmacocinétiques du lomitapide. Le lomitapide n'a pas été étudié chez des patients âgés de 65 ans ou plus.
Origine ethnique
Aucun ajustement de la dose n'est nécessaire pour les patients caucasiens ou latino-américains. Il n'y a pas suffisamment d'informations pour déterminer si la dose de Lojuxta nécessite d'être ajustée chez les personnes d'une autre origine. Cependant, comme le médicament est administré avec des doses croissantes en fonction de la sécurité individuelle des patients et de leur tolérance, aucune adaptation de la posologie est recommandée en fonction de l'origine ethnique.
Insuffisance rénale
Dans la population des patients insuffisants rénaux, le lomitapide n'a été étudié que chez des patients en insuffisance rénale au stade terminal (IRST). Une étude de pharmacocinétique chez des patients en insuffisance terminale sous hémodialyse a démontré une augmentation de 36 % de la concentration plasmatique moyenne de lomitapide, par rapport à celle de témoins sains appariés. La demi-vie terminale du lomitapide n'a pas été modifiée.
Insuffisance hépatique
Une étude en ouvert à dose unique a été menée afin d'évaluer les propriétés pharmacocinétiques de 60 mg de lomitapide chez des volontaires sains ayant une fonction hépatique normale, par rapport aux patients présentant une insuffisance hépatique légère (Child-Pugh A) et modérée (Child-Pugh B). Chez les patients insuffisants hépatiques modérés, l'ASC et la Cmax du lomitapide étaient plus élevées, respectivement de 164 % et 361 %, que celles des volontaires sains. Chez les patients insuffisants hépatiques légers, l'ASC et la Cmax du lomitapide étaient plus élevées, respectivement de 47 % et 4 %, que celles des volontaires sains. Lojuxta n'a pas été étudié chez des patients insuffisants hépatiques sévères (Child-Pugh score 10 à 15).
Population pédiatrique
Le lomitapide n'a pas été étudié chez des enfants âgés de moins de 18 ans.
Lojuxta a une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines.
Dans des études de toxicologie à doses orales répétées, menées chez des rongeurs et des chiens, les principaux résultats associés au médicament étaient une accumulation de lipides dans l'intestin grêle et/ou le foie, associée à des diminutions des taux sériques de cholestérol et/ou de triglycérides. Ces modifications sont secondaires au mécanisme d'action du lomitapide. D'autres modifications hépatiques observées dans des études de toxicité à doses répétées, menées chez des rats et des chiens comprenaient une augmentation des aminotransférases sériques, une inflammation subaiguë (seulement chez les rats) et une nécrose monocellulaire. Dans une étude d'un an à doses répétées chez des chiens, il n'y a pas eu de modifications microscopiques au niveau du foie, malgré une augmentation minime de l'ASAT sérique chez les femelles.
Une histiocytose pulmonaire a été observée chez les rongeurs. Les chiens présentaient une diminution des globules rouges, ainsi qu'une poikilocytose et/ou une anisocytose. Une toxicité testiculaire a été observée chez les chiens lors d'une exposition 205 fois plus forte que l'exposition humaine (ASC) avec 60 mg, dans une étude de 6 mois. Aucun effet indésirable sur les testicules n'a été observé dans une étude d'un an chez le chien pour une exposition de 64 fois celle atteinte chez l'homme avec 60 mg.
Dans une étude de carcinogénicité alimentaire menée chez des souris, le lomitapide a été administré jusqu'à 104 semaines, à des doses se situant entre 0,3 et 45 mg/kg/jour. Il a été observé des augmentations statistiquement significatives des incidences d'adénome et de carcinome hépatique à des doses ≥ à 1,5 mg/kg/jour chez les mâles (≥ à 2 fois l'exposition humaine avec 60 mg par jour sur la base de l'ASC) et ≥ à 7,5 mg/kg/jour chez les femelles (≥ à 9 fois l'exposition humaine avec 60 mg par jour sur la base de l'ASC). Les incidences de carcinome de l'intestin grêle et/ou d'adénome et de carcinome associés (tumeurs rares chez les souris) étaient augmentées de façon significative pour des doses ≥ à 15 mg/kg/jour chez les mâles (≥ à 26 fois l'exposition humaine pour 60 mg par jour sur la base de l'ASC) et de 15 mg/kg/jour chez les femelles (22 fois l'exposition humaine avec 60 mg par jour sur la base de l'ASC)).
Dans une étude de carcinogénicité orale menée chez des rats, le lomitapide a été administré jusqu'à 99 semaines à des doses allant jusqu'à 7,5 mg/kg/jour chez les mâles et 2,0 mg/kg/jour chez les femelles. Il a été observé une fibrose focale du foie chez les mâles et les femelles et une dégénérescence cystique hépatique uniquement chez les mâles. Chez les mâles ayant reçu de fortes doses, une augmentation de l'incidence de l'adénome pancréatique à cellules acineuses a été observée pour une exposition de 6 fois celle atteinte chez l'être humain avec 60 mg sur la base de l'ASC.
Le lomitapide n'était pas mutagène ni génotoxique dans toute une série d'études in vitro et in vivo.
Le lomitapide n'avait pas d'effet sur la reproduction chez les rats femelles à des doses allant jusqu'à 1 mg/kg, ni chez les rats mâles à des doses allant jusqu'à 5 mg/kg. Il a été estimé que les expositions systémiques au lomitapide à ces doses étaient 4 fois (chez les femelles) et 5 fois (chez les mâles) plus élevées que l'exposition humaine avec 60 mg sur la base de l'ASC.
Le lomitapide était tératogène chez les rats femelles en l'absence de toxicité maternelle pour une exposition (ASC) estimée deux fois plus élevée que celle atteinte chez l'être humain avec 60 mg. Il n'y avait pas de signe de toxicité embryofoetale chez les lapines pour une dose égale à 3 fois la dose humaine maximale recommandée (DHMR) de 60 mg sur la base de la surface corporelle. Une toxicité embryofoetale a été observée chez les lapines en l'absence de toxicité maternelle pour une dose ≥ à 6,5 fois la DHMR. Chez les furets, le lomitapide présentait à la fois une toxicité maternelle et une tératogénicité pour une dose < à 1 fois la DHMR.
Pas d'exigences particulières.
Liste I
Médicament nécessitant une surveillance particulière pendant le traitement
Prescription hospitalière
Prescription réservée aux spécialistes et services CARDIOLOGIE
Prescription réservée aux spécialistes et services DIABETOLOGIE
Prescription réservée aux spécialistes et services ENDOCRINOLOGIE
Prescription réservée aux spécialistes et services MALADIES METABOLIQUES
Prescription réservée aux spécialistes et services MEDECINE INTERNE
Gélule.
La gélule de 19,4 mm est constituée d'une coiffe et d'un corps orange, portant les inscriptions à l'encre noire « 5 mg » sur le corps et « A733 » sur la coiffe.
Flacon en polyéthylène haute densité (HDPE) scellé par induction par un opercule en polyester/aluminium/carton et muni d'un bouchon à vis en polypropylène.
Taille de l'emballage :
28 gélules
Chaque gélule contient du mésylate de lomitapide équivalent à 5 mg de lomitapide.
Excipient à effet notoire
Chaque gélule contient 70,12 mg de lactose (sous forme de monohydrate) (voir rubrique Mises en garde spéciales et précautions d'emploi).
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Contenu de la gélule :
Amidon (de maïs) prégélatinisé
Carboxyméthylamidon sodique (Type A)
Cellulose microcrystalline
Lactose monohydraté
Silice colloïdale anhydre
Stéarate de magnésium
Enveloppe de la gélule :
Gélatine
Dioxyde de titane (E171)
Oxyde de fer rouge (E172)
Encre d'impression:
Gomme laque
Oxyde de fer noir (E172)
Propylène glycol