
SARCLISA 20 mg-ml, solution à diluer pour perfusion, boîte de 1 flacon de 25 ml
Dernière révision : 17/06/2021
Taux de TVA : 2.1%
Laboratoire exploitant : SANOFI WINTHROP INDUSTRIE
Source :

SARCLISA est indiqué :
- en association avec le pomalidomide et la dexaméthasone pour le traitement des patients adultes atteints de myélome multiple en rechute et réfractaire, qui ont reçu au moins deux traitements antérieurs incluant le lénalidomide et un inhibiteur du protéasome et dont la maladie a progressé lors du dernier traitement.
- en association avec le carfilzomib et la dexaméthasone pour le traitement des patients adultes atteints de myélome multiple qui ont reçu au moins un traitement antérieur (voir rubrique Propriétés pharmacodynamiques).
Hypersensibilité à la substance active ou à l'un de ses excipients mentionnés à la rubrique Liste des excipients.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Réactions liées à la perfusion (RLP)
Des réactions liées à la perfusion, pour la plupart légères ou modérées, ont été observées chez 38,2 % des patients traités par SARCLISA dans l'étude ICARIA-MM et chez 45,8 % des patients traités par Isa-Kd dans l'étude IKEMA (voir rubrique Effets indésirables). Dans l'étude ICARIA-MM, toutes les réactions liées à la perfusion sont apparues au cours de la première perfusion de SARCLISA, et se sont résolues le jour- même pour 98 % des perfusions. Les symptômes les plus fréquents de RLP comprenaient dyspnée, toux, frissons et nausées. Les signes et symptômes sévères les plus fréquents incluaient hypertension, dyspnée et bronchospasme. Dans l'étude IKEMA, les réactions liées à la perfusion sont apparues le jour de la perfusion pour 99,2 % des épisodes. Chez les patients traités par Isa-Kd, 94,4 % de ceux ayant présenté une RLP l'ont présentée au cours du premier cycle de traitement. Toutes les réactions liées à la perfusion se sont résolues. Les symptômes les plus fréquents de réaction liée à la perfusion comprenaient toux, dyspnée, congestion nasale, vomissements et nausées. Les signes et les symptômes sévères les plus fréquents incluaient hypertension et dyspnée (voir rubrique Effets indésirables).
Cependant, des réactions graves liées à la perfusion incluant des réactions anaphylactiques sévères ont également été rapportées après l'administration de SARCLISA.
Pour diminuer les risques et la gravité des réactions liées à la perfusion, les patients doivent recevoir une prémédication par paracétamol, diphénhydramine ou équivalent avant la perfusion de SARCLISA ; la dexaméthasone doit être utilisée à la fois en tant que prémédication et comme traitement anti-myélome (voir rubrique Posologie et mode d'administration). Les signes vitaux doivent être fréquemment surveillés pendant toute la durée de la perfusion de SARCLISA. Lorsque nécessaire, interrompre la perfusion de SARCLISA et apporter des mesures médicales et de soutien adaptées (voir rubrique Posologie et mode d'administration). En cas de non-amélioration des symptômes à un grade ≤ 1 après l'interruption de la perfusion de SARCLISA, de persistance ou d'aggravation malgré l'utilisation de médicaments adaptés, de nécessité d'hospitalisation ou de menace du pronostic vital, arrêter le traitement par SARCLISA de manière définitive et mettre en place une prise en charge adaptée.
Neutropénie
Chez les patients traités par Isa-Pd, des cas de neutropénie ont été signalés comme des anomalies biologiques chez 96,1 % des patients et comme des effets indésirables (1) chez 46,7 % des patients, avec des cas de neutropénie de grade 3-4 signalés comme des anomalies biologiques chez 84,9 % des patients et comme des effets indésirables chez 45,4 % des patients. Des complications neutropéniques ont été observées chez 30,3 % des patients, dont 11,8 % de neutropénies fébriles et 25,0 % d'infections neutropéniques. Chez les patients traités par Isa-Kd, des cas de neutropénie ont été signalés comme des anomalies biologiques chez 54,8 % des patients et comme des effets indésirables(1) chez 4,5 % des patients, avec des cas de neutropénie de grade 3-4 signalés comme des anomalies biologiques chez 19,2 % des patients (dont 17,5 % de grade 3 et 1,7 % de grade 4) et comme des effets indésirables chez 4,0 % des patients. Des complications neutropéniques ont été observées chez 2,8 % des patients, dont 1,1 % de neutropénies fébriles et 1,7 % d'infections neutropéniques (voir rubrique Effets indésirables).
La numération formule sanguine doit être surveillée périodiquement pendant le traitement. Les patients atteints de neutropénie doivent faire l'objet d'une surveillance de tout signe d'infection. Aucune diminution de la dose de SARCLISA n'est recommandée. Il convient d'envisager des reports de doses de SARCLISA et l'utilisation de facteurs de croissance (par ex., G-CSF) pour diminuer le risque de neutropénie (voir rubrique Posologie et mode d'administration).
(1) Les valeurs hématologiques de laboratoire étaient enregistrées comme effets indésirables seulement si elles conduisaient à un arrêt de traitement et/ou une modification de la dose et/ou remplissaient un critère de gravité.
Infection
Une incidence plus élevée d'infections, y compris des infections de grade ≥ 3, principalement des pneumonies, des infections des voies aériennes supérieures et des bronchites, a été observée avec SARCLISA (voir rubrique Effets indésirables). Les patients recevant SARCLISA doivent être étroitement surveillés afin de détecter tout signe d'infection et un traitement standard approprié doit être instauré. Une prophylaxie antibiotique et antivirale peut être envisagée pendant le traitement.
Cancers secondaires
Dans l'étude ICARIA-MM, des cancers secondaires ont été rapportés chez 6 patients (3,9 %) traités par Isa-Pd et chez 1 patient (0,7 %) traité par Pd, et comprenaient un cancer de la peau chez 4 patients traités par Isa-Pd et chez 1 patient traité par Pd (voir rubrique Effets indésirables). Les patients ont poursuivi le traitement après résection du cancer de la peau. Dans l'étude IKEMA, des cancers secondaires ont été rapportés chez 13 patients (7,3 %) traités par Isa-Kd et chez 6 patients (4,9 %) traités par Kd. Les cancers secondaires étaient des cancers de la peau chez 9 patients (5,1 %) traités par Isa-Kd et chez 3 patients (2,5 %) traités par Kd, et étaient des tumeurs solides autres que le cancer de la peau chez 5 patients (2,8 %) traités par Isa-Kd et chez 4 patients (3,3 %) traités par Kd. Un patient (0,6 %) du groupe Isa-Kd et un patient (0,8 %) du groupe Kd présentaient à la fois un cancer de la peau et destumeurs solides autres que le cancer de la peau (voir rubrique Effets indésirables). Les patients atteints d'un cancer de la peau ont poursuivi le traitement après résection du cancer de la peau. Des tumeurs solides autres que le cancer de la peau ont été diagnostiquées dans les 3 mois suivant l'instauration du traitement chez 3 patients (1,7 %) traités par Isa-Kd et chez 2 patients (1,6 %) traités par Kd. L'incidence globale des cancers secondaires chez tous les patients exposés au SARCLISA est de 3,6 %. Les médecins doivent soigneusement évaluer les patients avant et pendant le traitement conformément aux directives de l'IMWG (International Myeloma Working Group) pour la survenue d'un cancer secondaire et initier le traitement indiqué.
Interférence avec les tests sérologiques (test indirect à l'antiglobuline)
SARCLISA se lie au CD38 sur les globules rouges (GR) et peut entraîner un faux positif au test indirect à l'antiglobuline (test de Coombs indirect). Afin d'éviter d'éventuels problèmes avec la transfusion de GR, chez les patients traités par SARCLISA, le groupe sanguin doit être déterminé et des tests de dépistage effectués avant la première perfusion. Le phénotypage peut être envisagé avant de commencer le traitement par SARCLISA selon la pratique locale. Si le traitement par SARCLISA a déjà commencé, le centre de transfusion sanguine doit être informé. Les patients doivent être surveillés pour prévenir le risque théorique d'hémolyse. Si une transfusion d'urgence est nécessaire, des concentrés de globules rouges ABO/RhD compatibles, sans épreuve directe de compatibilité, peuvent être administrés, conformément aux pratiques locales des établissements de transfusion sanguine (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). Il n'existe actuellement aucune information disponible concernant la durée de l'interférence avec le test indirect de Coombs après la dernière perfusion de SARCLISA. Selon les données relatives à la demi-vie d'isatuximab, il est anticipé qu'un test indirect de Coombs positif médié par isatuximab puisse persister environ 6 mois après la dernière perfusion.
Interférence avec la détermination de la réponse complète
L'isatuximab est un anticorps monoclonal IgG kappa qui peut être détecté sur l'électrophorèse des protéines sériques (EPS) et de l'immunofixations (IFE) utilisées pour la surveillance clinique de la protéine M endogène (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). Cette interférence peut avoir un impact sur l'exactitude de la détermination de la réponse complète chez certains patients ayant un myélome à IgG kappa. Vingt-deux patients dans le bras Isa-Pd qui remplissaient les critères de Très Bonne Réponse Partielle (TBRP) avec seulement une immunofixation positive résiduelle ont été soumis à un test d'interférence. Les échantillons de sérum provenant de ces patients ont été testés par spectrométrie de masse pour séparer le signal d'isatuximab du signal de la protéine M du myélome. Dans le bras Isa-Kd, sur les 27 patients identifiés comme présentant une interférence potentielle et testés par spectrométrie de masse au niveau de sensibilité du test d'immunofixation (25 mg/dl), 15 patients présentant une réponse non complète (non-RC) selon le comité de réponse indépendant (Independent Review Committee, IRC) n'ont montré aucune protéine M du myélome résiduelle détectable. Parmi ces 15 patients, 11 patients présentaient un taux de plasmocytes < 5 % dans la moelle osseuse. Cela indique que 11 patients supplémentaires sur les 179 patients du bras Isa-Kd (6,1 %) pourraient présenter une RC comme meilleure réponse, entraînant un taux de RC potentiel de 45,8 % (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Personnes âgées
Les données sont limitées chez les personnes âgées de ≥ 85 ans (voir rubrique Posologie et mode d'administration).
Résumé du profil de sécurité
Dans l'étude ICARIA-MM, les effets indésirables les plus fréquents (chez ≥ 20 % des patients) sont la neutropénie (46,7 %), les réactions liées à la perfusion (38,2 %), la pneumonie (30,9 %), l'infection des voies aériennes supérieures (28,3 %), la diarrhée (25,7 %) et la bronchite (23,7 %). Des effets indésirables graves sont survenus chez 61,8 % des patients recevant Isa-Pd. Les effets indésirables graves les plus fréquents sont la pneumonie (25,7 %) et la neutropénie fébrile (6,6 %). Un arrêt définitif du traitement en raison d'effets indésirables a été rapporté chez 7,2 % des patients traités par Isa-Pd. Des effets indésirables d'issue fatale au cours du traitement ont été rapportés chez 7,9 % des patients traités par Isa-Pd (ceux survenus chez plus de 1 % des patients étaient la pneumonie, survenue chez 1,3 % des patients, et d'autres infections, survenues chez 2,0 % des patients).
Dans l'étude IKEMA, les effets indésirables les plus fréquents (chez ≥ 20 % des patients) sont les réactions liées à la perfusion (45,8 %), l'hypertension (36,7 %), la diarrhée (36,2 %), l'infection des voies aériennes supérieures (36,2 %), la pneumonie (28,8 %), la fatigue (28,2 %), la dyspnée (27,7 %), l'insomnie (23,7 %), la bronchite (22,6 %) et la douleur dorsale (22,0 %). Des effets indésirables graves sont survenus chez 59,3 % des patients recevant Isa-Kd. L'effet indésirable grave le plus fréquent est la pneumonie (21,5 %). Un arrêt définitif du traitement en raison d'effets indésirables a été rapporté chez 8,5 % des patients traités par Isa-Kd. Des effets indésirables à d'issue fatale au cours du traitement ont été rapportés chez 3,4 % des patients traités par Isa-Kd (ceux survenus chez plus de 1 % des patients étaient la pneumonie et l'insuffisance cardiaque, toutes deux survenues chez 1,1 % des patients).
Tableau des effets indésirables
Les effets indésirables ont été décrits à l'aide des Critères Communs de Toxicité du NCI, des termes du COSTART et de MedDRA. Les fréquences sont définies ainsi : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de gravité.
Les effets indésirables ont été rapportés chez les 152 patients ayant reçu Isa-Pd avec une durée médiane d'exposition de 41 semaines dans l'étude ICARIA-MM (voir rubrique Propriétés pharmacodynamiques).
Tableau 3a - Effets indésirables rapportés chez des patients présentant un myélome multiple traités par isatuximab en association avec du pomalidomide et de la dexaméthasone (ICARIA-MM)
| Classe de systèmes d'organes | | Fréquence | Incidence (%) |
| Terme préférentiel | Effets indésirables | | (N=152) | |
| Tous grades | Grade ≥3 | |||
| Infections et infestations | Pneumonieb c | Très fréquent | 47 (30.9) | 40 (26.3) |
| Infection des voies aériennes supérieures* | Très fréquent | 43 (28.3) | 5 (3.3) | |
| Bronchite* | Très fréquent | 36 (23.7) | 5 (3.3) | |
| Tumeurs bénignes et malignes (incluant kystes et polypes) | Carcinome épidermoïde cutané | Fréquent | 4 (2.6) | 2 (1.3) |
| Affections hématologiques et du système lymphatique | Neutropénied | Très fréquent | 71 (46.7) | 70 (46.1) |
| Neutropénie fébrile | Très fréquent | 18 (11.8) | 18 (11.8) | |
| Affections du système immunitaire | Réaction anaphylactiquee | Peu fréquent | 5 (0.3) | 5 (0.3) |
| Troubles du métabolisme et de la nutrition | Diminution de l'appétit* | Fréquent | 15 (9.9) | 2 (1.3) |
| Affections cardiaques | Fibrillation auriculaire | Fréquent | 7 (4.6) | 3 (2.0) |
| Affections respiratoires, thoraciques et médiastinales | Dyspnée* | Très fréquent | 23 (15.1) | 6 (3.9) |
| Affections gastro-intestinales | Diarrhée* | Très fréquent | 39 (25.7) | 3 (2.0) |
| Nausées* | Très fréquent | 23 (15.1) | 0 | |
| Vomissements* | Très fréquent | 18 (11.8) | 2 (1.3) | |
| Investigations | Perte de poids* | Fréquent | 10 (6.6) | 0 |
| Lésion, intoxication et complications d'intervention | Réaction à la perfusionc | Très fréquent | 58 (38.2) | 4 (2.6) |
b Le terme pneumonie est un regroupement des termes suivants : pneumonie atypique, aspergillose broncho- pulmonaire, pneumonie, pneumonie à Haemophilus, pneumonie à Influenza, pneumonie à Pneumocoque, pneumonie à Streptocoque, pneumonie virale, pneumonie bactérienne, infection à Haemophilus, infection pulmonaire, pneumonie fongique, et pneumonie à Pneumocystis jirovecii.
c Voir « Description de certains effets indésirables »
d Les valeurs hématologiques de laboratoire étaient enregistrées comme TEAE seulement si elles conduisaient à un arrêt de traitement ou une modification de la dose et/ou remplissaient un critère de gravité.
e Basé sur les essais cliniques dans le myélome multiple
* pas de grade 4
Les effets indésirables ont été rapportés chez les 177 patients ayant reçu Isa-Kd avec une durée médiane d'exposition de 80,0 semaines dans l'étude IKEMA (voir rubrique Propriétés pharmacodynamiques).
Tableau 4a : Effets indésirables rapportés chez des patients présentant un myélome multiple traités par isatuximab en association avec du carfilzomib et de la dexaméthasone (Étude IKEMA)
| Classe de systèmes d'organes Terme préférentiel | Effets indésirables | Fréquence | Incidence (%) (N = 177) | |
| Tous grades | Grade ≥ 3 | |||
| Infections et infestations | Pneumoniebc | Très fréquent | 28,8 % | 20,9 |
| Infection des voies aériennes supérieures* | Très fréquent | 36,2 % | 3,4 % | |
| Bronchite* | Très fréquent | 22,6 % | 2,3 % | |
| Affections vasculaires | Hypertension* | Très fréquent | 36,7 % | 20,3 % |
| Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes) | Cancers de la peau* | Fréquent | 5,1 % | 0,6% |
| Tumeurs solides autres que le cancer de la peau | Fréquent | 3.4% | 1,7 % | |
| Affections hématologiques et du système lymphatique | Neutropénied | Fréquent | 4,5% | 4,0% |
| Affections du système immunitaire | Réaction anaphylactiquee | Peu fréquent | 5 (0,3 %) | 5 (0,3 %) |
| Affections respiratoires, thoraciques et médiastinales | Dyspnée* | Très fréquent | 27,7 % | 5,1 % |
| Toux* | Très fréquent | 19,8 % | 0 % | |
| Affections gastro- intestinales | Diarrhée* | Très fréquent | 36,2 % | 2,8% |
| Vomissements* | Très fréquent | 15,3 % | 1,1 % | |
| Troubles généraux et anomalies au site d'administration | Fatigue* | Très fréquent | 28,2 % | 3,4 % |
| Lésions, intoxications et complications liées aux procédures | Réaction à la perfusionc* | Très fréquent | 45,8 % | 0,6% |
b Le terme pneumonie est un regroupement des termes suivants : pneumonie atypique, pneumonie à Pneumocystis jirovecii, pneumonie, pneumonie grippale, pneumonie à légionelles, pneumonie streptococcique, pneumonie virale et sepsis pulmonaire.
c Voir « Description de certains effets indésirables »
d Les valeurs hématologiques de laboratoire étaient enregistrées comme TEAE seulement si elles conduisaient à un arrêt de traitement ou une modification de la dose ou remplissaient un critère de gravité.
e Basé sur les essais cliniques dans le myélome multiple
* Pas de grade 4 ou 5.
Description de certains effets indésirables
Réactions liées à la perfusion
Dans l'étude ICARIA-MM, des réactions liées à la perfusion ont été rapportées chez 58 patients (38,2 %) traités par SARCLISA. Tous les patients ayant présenté des réactions liées à la perfusion les ont présentés pendant la première perfusion de SARCLISA, avec 3 patients (2,0 %) ayant égalementprésenté des réactions liées à la perfusion lors de leur deuxième perfusion, et 2 patients (1,3 %) lors de leur quatrième perfusion. Des réactions liées à la perfusion de grade 1 ont été rapportées chez 3,9 % des patients, de grade 2 chez 31,6 %, de grade 3 chez 1,3 %, et de grade 4 chez 1,3 % des patients. Toutes les réactions liées à la perfusion étaient réversibles et ont été résolues le jour même dans 98 % des épisodes. Les signes et symptômes des réactions liées à la perfusion de grade 3 ou 4 incluaient la dyspnée, l'hypertension et les bronchospasmes.
L'incidence des interruptions de la perfusion dues à des réactions liées aux perfusions était de 28,9 %. Le délai médian jusqu'à l'interruption de la perfusion était de 55 minutes.
Des interruptions du traitement dues à une réaction à la perfusion ont été rapportées chez 2,6 % des patients dans le bras de traitement par Isa-Pd.
Dans l'étude IKEMA, des réactions liées à la perfusion ont été rapportées chez 81 patients (45,8 %) traités par Isa-Kd. Des réactions liées à la perfusion de grade 1 ont été rapportées chez 13,6 % des patients, de grade 2 chez 31,6 % et de grade 3 chez 0,6 % des patients traités par Isa-Kd. Toutes les réactions liées à la perfusion étaient réversibles ; elles ont été résolues le jour-même pour 73,8 % des épisodes chez les patients du bras Isa-Kd et en plus de 2 jours pour 2,5 % des épisodes chez les patients du bras Isa-Kd. Les signes et symptômes des réactions liées à la perfusion de grade 3 incluaient la dyspnée et l'hypertension. L'incidence des interruptions de la perfusion d'isatuximab dues à des réactions liées à la perfusion était de 29,9 %. Le délai médian jusqu'à l'interruption de la perfusion d'isatuximab était de 63 minutes. L'isatuximab a été arrêté chez 0,6 % des patients en raison de réactions liées à la perfusion (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Infections
Dans l'étude ICARIA-MM, l'incidence des infections de grade 3 ou plus était de 42,8 %. La pneumonie était l'infection sévère la plus fréquemment rapportée avec un grade 3 signalé chez 21,7 % des patients dans le groupe Isa-Pd comparé à 16,1 % dans le groupe Pd, et un grade 4 chez 3,3 % des patients dans le groupe Isa-Pd comparé à 2,7 % dans le groupe Pd. Des interruptions du traitement dues à une infection ont été rapportées chez 2,6 % des patients dans le groupe Isa-Pd comparé à 5,4 % dans le groupe Pd. Des infections d'issues fatales ont été rapportées chez 3,3 % des patients dans le groupe Isa-Pd et 4,0 % dans le groupe Pd. Dans l'étude IKEMA, l'incidence des infections de grade 3 ou plus était de 38,4 %. La pneumonie était l'infection sévère la plus fréquemment rapportée avec un grade 3 signalé chez 15,8 % des patients dans le groupe Isa-Kd comparé à 10,7 % dans le groupe Kd, et un grade 4 chez 3,4 % des patients dans le groupe Isa-Kd comparé à 2,5 % dans le groupe Kd. Le traitement a été interrompu en raison d'une infection chez 2,8 % des patients dans le groupe Isa-Kd comparé à 4,9 % dans le groupe Kd. Des infections d'issues fatales ont été rapportées chez 2,3 % des patients dans le groupe Isa-Kd et 0,8 % dans le groupe Kd (voir rubrique Mises en garde spéciales et précautions d'emploi).
Insuffisance cardiaque
Dans l'étude IKEMA, une insuffisance cardiaque (comprenant insuffisance cardiaque, insuffisance cardiaque congestive, insuffisance cardiaque aiguë, insuffisance cardiaque chronique, insuffisance ventriculaire gauche et œdème pulmonaire) a été rapportée chez 7,3 % des patients dans le groupe Isa- Kd (4,0 % de grade ≥ 3) et chez 6,6 % des patients dans le groupe Kd (4,1 % de grade ≥ 3). Une insuffisance cardiaque grave a été observée chez 4,0 % des patients dans le groupe Isa-Kd et chez 3,3 % des patients dans le groupe Kd. Une insuffisance cardiaque d'issue fatale sous traitement a été rapportée chez 1,1% des patients du groupe Isa-Kd, et aucune n'a été rapportée dans le groupe Kd (voir les informations de prescription actuelles pour le carfilzomib).
Valeurs hématologiques de laboratoire
Tableau 5 : Anomalies hématologiques chez les patients recevant isatuximab en association avec pomalidomide et dexaméthasone versus pomalidomide et dexaméthasone (ICARIA-MM)
| Paramètre biologique | SARCLISA + Pomalidomide + dexaméthasone n (%) (N = 152) | Pomalidomide + dexaméthasone n (%) (N = 147) | ||||
| Tous grades | Grade 3 | Grade 4 | Tous grades | Grade 3 | Grade 4 | |
| Anémie | 151 (99,3) | 48 (31,6) | 0 | 145 (98,6) | 41 (27,9) | 0 |
| Neutropénie | 146 (96,1) | 37 (24,3) | 92 (60,5) | 137 (93,2) | 57 (38,8) | 46 (31,3) |
| Lymphopénie | 140 (92,1) | 64 (42,1) | 19 (12,5) | 137 (93,2) | 52 (35,4) | 12 (8,2) |
| Thrombopénie | 127 (83,6) | 22 (14,5) | 25 (16,4) | 118 (80,3) | 14 (9,5) | 22 (15,0) |
Le dénominateur utilisé pour le calcul du pourcentage est le nombre de patients ayant été évalués au moins 1 fois (examen biologique) pendant la période d'observation considérée.
Tableau 6 : Anomalies hématologiques chez les patients recevant isatuximab en association avec carfilzomib et dexaméthasone versus carfilzomib et dexaméthasone (IKEMA)
| Paramètre biologique | SARCLISA + carfilzomib + dexaméthasone (N = 177) | Carfilzomib + dexaméthasone (N = 122) | ||||
| | Tous grades | Grade 3 | Grade 4 | Tous grades | Grade 3 | Grade 4 |
| Anémie | 99,4 % | 22,0 % | 0 % | 99,2 % | 19,7 % | 0 % |
| Neutropénie | 54,8 % | 17,5 % | 1,7 % | 43,4 % | 6,6 % | 0,8 % |
| Lymphopénie | 94,4 % | 52,0 % | 16,9 % | 95,1 % | 43,4 % | 13,9 % |
| Thrombopénie | 94,4 % | 18,6 % | 11,3 % | 87,7 % | 15.6% | 8,2 % |
Le dénominateur utilisé pour le calcul du pourcentage est le nombre de patients ayant été évalués au moins 1 fois (examen biologique) pendant la période d'observation considérée.
Immunogénicité
Dans 9 études cliniques sur le myélome multiple (MM) avec des traitements par isatuximab en monothérapie et en association, y compris ICARIA-MM et IKEMA (n = 1018), l'incidence du développement d'anticorps anti-médicament (AAM) après introduction du traitement était de 1,9 %. Aucun effet des AAM n'a été observé sur la pharmacocinétique, la sécurité d'emploi ou l'efficacité d'isatuximab.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
Une prémédication doit être mise en place 15 à 60 minutes avant de
démarrer la perfusion afin de réduire les risques et la gravité des réactions à la perfusion.
Afin d'éviter d'éventuels problèmes avec la transfusion de GR, chez les
patients traités, le groupe sanguin doit être déterminé et
des tests de dépistage effectués avant la première perfusion. Le
phénotypage peut être envisagé avant de commencer le traitement selon la pratique locale.
SURVEILLANCE :
- signes vitaux
- numération sanguine
- signe d'infection.
INTERFERENCE AVEC LA DETERMINATION DE LA REPONSE
:
Le traitement est un anticorps monoclonal IgG kappa qui peut être détecté de manière fortuite par
l'électrophorèse des protéines sériques (EPS) et de l'immunofixations (IFE) utilisés pour la
surveillance clinique de la protéine M endogène. Cette interférence peut avoir un
impact sur l'exactitude de la détermination de la réponse complète chez certains patients ayant un
myélome à IgG kappa.
INTERFERENCE AVEC LES ANALYSES SEROLOGIQUES :
Comme la protéine CD38 s'exprime à la surface des
globules rouges, le traitement, un anticorps anti- CD38, peut interférer avec les
tests sérologiques des centres de transfusion sanguine en produisant
potentiellement des réactions de faux positif aux tests indirects à
l'antiglobuline (tests de Coombs indirect), tests de détection d'anticorps
(dépistage), des panels d'identification des anticorps et tests anti «
immunoglobuline-humaine » (AHG).
INTERFERENCE avec les tests d'électrophorèse de
protéine sérique et d'immunofixation :
Le traitement peut être détecté de manière fortuite par les tests d'électrophorèse de protéines sériques (EPS) et d'immunofixation (IFE) utilisés pour surveiller la protéine M, et pourrait interférer avec une classification exacte de la réponse basée selon les critères de l' « International Myeloma Working Group » (IMWG).
Il n'existe actuellement aucune information disponible concernant la durée de l'interférence avec le test indirect de Coombs après la dernière perfusion. Selon les données relatives à la demi-vie d'isatuximab, il est anticipé qu'un test indirect de Coombs positif médié par isatuximab puisse persister environ 6 mois après la dernière perfusion.Les femmes en âge de procréer doivent utiliser une méthode de contraception efficace pendant le traitement et pendant au moins 5 mois après l'arrêt du traitement.
Femmes en âge de procréer/Contraception
Les
femmes en âge de procréer traitées par isatuximab doivent utiliser une
méthode de contraception efficace pendant le traitement et pendant 5
mois après l'arrêt du traitement.
Grossesse
On ne dispose d‘aucune donnée sur l'utilisation d'isatuximab chez la femme enceinte. Aucune étude de toxicité sur la reproduction animale n'a été menée avec isatuximab. Les anticorps monoclonaux IgG1 sont connus pour traverser le placenta après le premier trimestre de grossesse. L'utilisation d'isatuximab chez la femme enceinte n'est pas recommandée.
Allaitement
Aucune donnée n'est disponible concernant l'excrétion d'isatuximab dans le lait maternel. Les IgG humaines sont connues pour être excrétées dans le lait maternel au cours des premiers jours suivant la naissance et leurs taux diminuent jusqu'à de faibles concentrations peu après ; cependant, le risque pour l'enfant allaité ne peut pas être exclu pendant cette courte période juste après la naissance.Pendant cette période spécifique, il convient de décider si l'allaitement doit être interrompu ou si le traitement par isatuximab doit être arrêté en tenant compte du bénéfice de l'allaitement pour l'enfant et du bénéfice du traitement pour la femme. Par la suite, isatuximab peut être utilisé pendant l'allaitement si cela est cliniquement nécessaire.
Fertilité
Il n'existe aucune donnée chez l'homme ni chez l'animal pour déterminer les effets potentiels d'isatuximab sur la fertilité chez l'homme et la femme (voir rubrique Données de sécurité précliniques).
Pour les autres médicaments administrés avec SARCLISA, veuillez consulter le résumé actuel des caractéristiques du produit correspondant.
L'isatuximab n'a aucun impact sur la pharmacocinétique du pomalidomide ou du carfilzomib, ou vice versa.
Interférence avec les analyses sérologiques
Comme la protéine CD38 s'exprime à la surface des globules rouges, isatuximab, un anticorps anti- CD38, peut interférer avec les tests sérologiques des centres de transfusion sanguine en induisant potentiellement des résultats faux positif aux tests indirects à l'antiglobuline (tests de Coombs indirect), aux tests de détection d'anticorps (dépistage), aux panels d'identification des anticorps et aux tests anti« immunoglobuline-humaine » (AGH) chez les patients traités par isatuximab (voir rubrique Mises en garde spéciales et précautions d'emploi).
Les méthodes d'atténuation de l'interférence comprennent le traitement des panels de globules rouges par du dithiothréitol (DTT) pour perturber la liaison avec isatuximab ou toutes autres méthodes validées localement. Le système de groupe sanguin Kell étant également sensible au traitement par DTT, des unités Kell négatives doivent être fournies après avoir exclu ou identifié la présence d'allo- anticorps en utilisant des GR traités par DTT.
Interférence avec les tests d'électrophorèse de protéine sérique et d'immunofixation
L'isatuximab peut être détecté par les tests d'électrophorèse de protéines sériques (EPS) et d'immunofixation (IFE) utilisés pour surveiller la protéine M, et pourrait interférer avec une classification exacte de la réponse basée selon les critères de l'« International Myeloma Working Group » (IMWG) (voir rubrique Mises en garde spéciales et précautions d'emploi).
SARCLISA doit être administré par un professionnel de santé, dans un environnement où des dispositifs de réanimation sont disponibles.
Prémédication
Une prémédication doit être mise en place avant la perfusion de SARCLISA avec les médicaments suivants afin de réduire le risque et la gravité des réactions liées à la perfusion :
Dexaméthasone 40 mg par voie orale ou intraveineuse (ou 20 mg par voie orale ou intraveineuse pour les patients ≥ 75 ans) lors de l'administration en association avec isatuximab et pomalidomide,
Dexaméthasone 20 mg (par voie intraveineuse les jours de perfusion d'isatuximab et/ou de carfilzomib, et par voie orale les autres jours) lors de l'administration en association avec isatuximab et carfilzomib.
Paracétamol 650 mg à 1000 mg par voie orale (ou équivalent).
Diphénhydramine 25 mg à 50 mg par voie intraveineuse ou orale (ou équivalent [par ex. cétirizine, prométhazine, dexchlorphéniramine]). La voie intraveineuse est privilégiée, au moins en ce qui concerne les 4 premières perfusions.
Les médicaments de prémédication recommandés doivent être administrés 15 à 60 minutes avant de démarrer la perfusion de SARCLISA. Les besoins en matière de prémédication pourront être reconsidérés pour les patients qui ne présentent pas de réaction à la perfusion lors des 4 premières administrations de SARCLISA.
Gestion de la neutropénie
L'utilisation de facteurs stimulant les colonies (par ex. G-CSF) doit être envisagée pour diminuer le risque de neutropénie. En cas de présence d'une neutropénie de grade 4, l'administration de SARCLISA doit être reportée jusqu'à ce que la numération des neutrophiles revienne à un taux d'au moins 1,0 x 109/l. (voir rubrique Mises en garde spéciales et précautions d'emploi).
PosologieLa dose recommandée de SARCLISA est de 10 mg/kg de poids corporel, administrée par perfusion intraveineuse en association avec le pomalidomide et la dexaméthasone (Isa-Pd) ou en association avec le carfilzomib et la dexaméthasone (Isa-Kd), conformément au calendrier d'administration présenté dans le tableau 1 :
Tableau 1 : Schéma posologique de SARCLISA en association avec le pomalidomide et la dexaméthasone ou en association avec le carfilzomib et la dexaméthasone
| Cycles | Schéma posologique |
| Cycle 1 | Jours 1, 8, 15 et 22 (hebdomadaire) |
| Cycle 2 et suivants | Jours 1 et 15 (toutes les 2 semaines) |
Chaque cycle de traitement correspond à une période de 28 jours. Le traitement est renouvelé jusqu'à progression de la maladie ou survenue d'une toxicité inacceptable.
Pour les autres médicaments qui sont administrés avec SARCLISA, voir la rubrique Propriétés pharmacodynamiques et le résumé des caractéristiques des produits correspondants en vigueur.
Le calendrier d'administration doit être respecté scrupuleusement. En cas d'oubli d'une dose prévue de SARCLISA, administrer la dose dès que possible et ajuster le schéma thérapeutique en conséquence, afin de maintenir l'intervalle de traitement.
Ajustements de la posologie
Aucune diminution de dose de SARCLISA n'est recommandée.
Des ajustements au niveau de l'administration doivent être effectués si les patients présentent des réactions liées à la perfusion (voir « Mode d'administration » ci-dessous).
En ce qui concerne les autres médicaments administrés avec SARCLISA, se reporter au Résumé des Caractéristiques du Produit correspondant en vigueur.
Populations particulières
Personnes âgées
D'après l'analyse de pharmacocinétique de population, aucun ajustement posologique n'est recommandé pour les personnes âgées.
Patients avec une insuffisance rénale
D'après l'analyse pharmacocinétique de population et de sécurité clinique, aucun ajustement posologique n'est recommandé chez les patients présentant une insuffisance rénale légère à sévère (voir rubrique Propriétés pharmacocinétiques).
Patients avec une insuffisance hépatique
D'après l'analyse pharmacocinétique de population, aucun ajustement posologique n'est recommandé chez les patients présentant une insuffisance hépatique légère. Les données disponibles sont limitées chez les patients atteints d'une insuffisance hépatique modérée et sévère (voir rubrique Propriétés pharmacocinétiques), mais aucune donnée probante ne tend à suggérer qu'un ajustement de la posologie est requis chez ces patients.
Population pédiatrique
La sécurité et l'efficacité de SARCLISA chez les enfants de moins de 18 ans n'ont pas été établies. Aucune donnée n'est disponible.
Mode d'administration
SARCLISA est destiné à un usage intraveineux. Pour les instructions concernant la dilution du médicament avant administration, voir la rubrique Précautions particulières d'élimination et de manipulation.
Débits de perfusion
Après la dilution, la perfusion de SARCLISA doit être administrée par voie intraveineuse aux débits présentés dans le tableau 2 (voir rubrique Propriétés pharmacodynamiques). L'augmentation progressive du débit de perfusion ne peut être envisagée qu'en absence de réactions liées à la perfusion (voir rubrique Effets indésirables).
Tableau 2 : Débits de perfusion lors de l'administration de SARCLISA
| | Volume de dilution | Débit initial | Absence de réaction à la perfusion | Augmentation du débit | Débit maximum |
| Première perfusion | 250 ml | 25 ml/heure | Pendant 60 minutes | 25 ml/heure toutes les 30 minutes | 150 ml/heure |
| Deuxième perfusion | 250 ml | 50 ml/heure | Pendant 30 minutes | 50 ml/heure pendant 30 minutes, puis augmentation de 100 ml/heure | 200 ml/heure |
| Perfusions suivantes | 250 ml | 200 ml/heure | ------- | ------- | 200 ml/heure |
Des ajustements d'administration doivent être effectués si les patients présentent des réactions liées à la perfusion (voir rubrique Mises en garde spéciales et précautions d'emploi).
Chez les patients ayant besoin d'un ajustement (réactions liées à la perfusion modérées, de grade 2), une interruption temporaire de la perfusion doit être envisagée et un traitement symptomatique supplémentaire peut être administré. Après amélioration des symptômes à un grade ≤ 1 (réactions légères), la perfusion de SARCLISA pourra être reprise à la moitié du débit initial sous surveillance étroite et avec des soins de support si nécessaire. Si les symptômes ne réapparaissent pas après 30 minutes, le débit de perfusion pourra être augmenté jusqu'au débit initial, puis augmenté progressivement comme indiqué dans le tableau 2.
Si les symptômes ne disparaissent pas rapidement ou ne s'améliorent pas à un grade ≤ 1 après l'interruption de la perfusion de SARCLISA, s'ils persistent ou s'aggravent malgré un traitement adapté, nécessitent une hospitalisation ou menacent le pronostic vital, le traitement par SARCLISA doit être définitivement arrêté et un traitement de support supplémentaire doit être administré si nécessaire.
Durée de conservation :
Flacon non ouvert
3 ans.
Après dilution
La stabilité chimique et physique de la solution de perfusion de SARCLISA a été démontrée pendant 48 heures entre 2 °C et 8 °C, puis pendant 8 heures (y compris le temps de perfusion) à température ambiante (entre 15 °C et 25 °C).
D'un point de vue microbiologique, le produit doit être utilisé immédiatement. S'il n'est pas utilisé immédiatement, la durée et les conditions de conservation sont sous la responsabilité de l'utilisateur et ne devraient pas dépasser 24 heures à une température comprise entre 2 °C et 8 °C, sauf si la dilution a eu lieu dans des conditions aseptiques contrôlées et validées.
La poche de perfusion ne nécessite aucune protection contre la lumière.
Précautions particulières de conservation :
A conserver au réfrigérateur (entre 2°C et 8°C). Ne pas congeler.
À conserver dans l'emballage d'origine à l'abri de la lumière.
Pour les conditions de conservation du médicament après dilution, voir la rubrique Durée de conservation.
Ce médicament ne doit pas être mélangé avec d'autres médicaments à l'exception de ceux mentionnés dans la rubrique Précautions particulières d'élimination et de manipulation.
Signes et symptômes
Il n'y a eu aucun cas de surdosage d'isatuximab lors des études cliniques. Des doses jusqu'à 20 mg/kg d'isatuximab par voie intraveineuse ont été administrées lors des études cliniques.
Prise en charge
Il n'existe aucun antidote connu spécifique à utiliser en cas de surdosage avec SARCLISA. En cas de surdosage, surveiller les patients pour détecter tout signe ou symptôme d'effets indésirables et prendre toutes les mesures adaptées immédiatement.
Classe pharmacothérapeutique : Antinéoplasiques, anticorps monoclonaaux, code ATC : L01XC38.
Mécanisme d'action
L'isatuximab est un anticorps monoclonal de type IgG1 qui se lie à un épitope extracellulaire spécifique du récepteur CD38. Le CD38 est une glycoprotéine transmembranaire fortement exprimée sur les cellules du myélome multiple.
In vitro, isatuximab agit par des mécanismes dépendant du Fc de l'IgG, notamment la cytotoxicité à médiation cellulaire dépendante des anticorps (antibody dependent cell mediated cytotoxicity, ADCC), la phagocytose cellulaire dépendante des anticorps (antibody dependent cellular phagocytosis, ADCP) et la cytotoxicité dépendante des compléments (CDC). De plus, isatuximab peut aussi déclencher la mort des cellules tumorales par l'induction de l'apoptose via un mécanisme indépendant du Fc.
In vitro, isatuximab bloque l'activité enzymatique du CD38 qui catalyse la synthèse et l'hydrolyse de l'ADP-ribose cyclique, un agent mobilisant le calcium. L'isatuximab inhibe la production d'ADPRc à partir de Nicotinamide Adénine Dinucléotide (NAD) extracellulaire dans les cellules du myélome multiple.
In vitro, isatuximab peut activer les cellules NK (Natural killer) en l'absence de cellules tumorales CD38 cibles positives.
In vivo, une diminution de la numération absolue des cellules NK totales CD16+ et CD56+, des lymphocytes B CD19+, des lymphocytes T CD4+ et des TREG (CD3+, CD4+, CD25+, CD127-) a été observée dans le sang périphérique des patients traités par isatuximab en monothérapie.
Chez les patients atteints de myélome multiple, SARCLISA en monothérapie a induit une expansion clonale du répertoire des récepteurs des lymphocytes T, indiquant une réponse immunitaire adaptative.
L'association in vitro d'isatuximab et de pomalidomide améliore, par rapport à isatuximab seul, la lyse des cellules myélomateuses exprimant le CD38 par le biais de cellules effectrices (ADCC) et par la destruction directe des cellules tumorales. Les expériences in vivo chez l'animal utilisant un modèle de xénogreffe de myélome multiple humain chez la souris ont démontré que l'association d'isatuximab et de pomalidomide entraînait une amélioration de l'activité antitumorale par rapport à l'activité d'isatuximab ou du pomalidomide seul.
Efficacité et sécurité cliniques
ICARIA-MM (EFC14335)
L'efficacité et la sécurité d'emploi de SARCLISA en association avec le pomalidomide et la dexaméthasone ont été évaluées dans l'étude ICARIA-MM (EFC14335), une étude de phase III multicentrique, internationale, randomisée, en ouvert, comparative en groupes parallèles, menée chez des patients atteints de myélome multiple en rechute et/ou réfractaire. Les patients avaient reçu au moins deux traitements antérieurs, dont le lénalidomide et un inhibiteur de protéasome, et présentaient une progression de la maladie sous traitement ou au plus tard 60 jours après la fin du traitement précédent. Les patients atteints d'une maladie réfractaire primaire étaient exclus.
Un total de 307 patients ont été randomisés selon un rapport de 1:1 pour recevoir soit SARCLISA en association avec du pomalidomide et de la dexaméthasone (Isa-Pd, 154 patients), soit du pomalidomide et de la dexaméthasone (Pd, 153 patients). Le traitement a été administré dans les deux groupes par cycles de 28 jours jusqu'à progression de la maladie ou toxicité inacceptable. SARCLISA 10 mg/kg a été administré par perfusion IV chaque semaine au cours du premier cycle puis toutes les deux semaines par la suite. Le pomalidomide 4 mg a été pris par voie orale une fois par jour du jour 1 au jour 21 de chaque cycle de 28 jours. La dexaméthasone (par voir orale/intraveineuse) 40 mg (20 mg pour les patients âgés de ≥ 75 ans) a été administrée les jours 1, 8, 15 et 22 de chaque cycle de 28 jours.
Dans l'ensemble, à l'inclusion les caractéristiques démographiques et celles de la maladie étaient similaires entre les deux bras de traitement, avec quelques différences mineures. L'âge médian des patients était de 67 ans (intervalle de 36 à 86 ans) ; 19,9 % des patients étaient âgés de ≥ 75 ans. L'indice de performance ECOG était de 0 chez 35,7 % des patients dans le bras isatuximab et 45,1 % dans le bras comparateur, de 1 chez 53,9 % des patients dans le bras isatuximab et 44,4 % dans le bras comparateur, et de 2 chez 10,4 % des patients dans le bras isatuximab et 10,5 % dans le bras comparateur, 10,4 % des patients dans le bras isatuximab contre 10,5 % dans le bras comparateur ont intégré l'étude avec des antécédents de BPCO ou d'asthme, et 38,6 % contre 33,3 % des patients présentant une atteinte de la fonction rénale (clairance de la créatinine < 60 ml/min/1,73 m²) ont été inclus dans le bras isatuximab par rapport au bras comparateur, respectivement. Selon le système de classification international (International Staging System, ISS), le stade de la maladie à l'entrée dans l'étude était de I chez 37,5 % (41,6 % dans le bras isatuximab et 33,3 % dans le bras comparateur), de II chez 35,5 % (34,4 % dans le bras isatuximab et 36,6 % dans le bras comparateur) et de III chez 25,1 % (22,1 % dans le bras isatuximab et 28,1 % dans le bras comparateur) des patients. Dans l'ensemble, 19,5 % des patients (15,6 % dans le bras isatuximab et 23,5 % dans le bras comparateur) présentaient des anormalités des chromosomes à haut risque à l'entrée dans l'étude ; del(17p), t(4;14) et t(14;16) étaient présents chez 12,1 % (9,1 % dans le bras isatuximab et 15,0 % dans le bras comparateur), 8,5 % (7,8 % dans le bras isatuximab et 9,2 % dans le bras comparateur) et 1,6 % (0,6 % dans le bras isatuximab et 2,6 % dans le bras comparateur) des patients, respectivement.
Le nombre médian de lignes antérieures de traitements était de 3 (intervalle de 2 à 11). Tous les patients avaient reçu au préalable un inhibiteur du protéasome et du lénalidomide, et 56,4 % des patients avaient fait l'objet d'une greffe de cellules souches. La majorité des patients (92,5 %) étaient réfractaires au lénalomide, 75,9 % à un inhibiteur du protéasome, 72,6 % à un immunomodulateur et à un inhibiteur du protéasome, et 59 % des patients étaient réfractaires au lénalidomide en traitement de dernière ligne.
La durée médiane de traitement était de 41 semaines pour le groupe Isa-Pd et de 24 semaines pour le groupe Pd.
La survie sans progression (SSP) était le critère d'évaluation principal de l'efficacité de l'étude ICARIA- MM. L'amélioration de SSP représentait une diminution de 40,4 % du risque de progression de la maladie ou de décès chez les patients traités par Isa-Pd.
Les résultats en matière d'efficacité sont présentés dans le Tableau 7 et les courbes de Kaplan-Meier pour la SSP et la SG sont représentées aux Figures 1 et 2 :
Tableau 7 : Efficacité de SARCLISA en association avec le pomalidomide et la dexaméthasone par rapport à l'association pomalidomide et dexaméthasone dans le traitement du myélome multiple (analyse de la population en intention de traiter)
| Critère d'évaluation | SARCLISA + pomalidomide + dexaméthasone N = 154 | Pomalidomide + dexaméthasone N = 153 |
| Survie sans progressiona b | | |
| Médiane (mois) [IC à 95 %] | 11,53 [8,936-13,897] | 6,47 [4,468-8,279] |
| Hazard Ratio c [IC à 95 %] | 0,596 [0,436-0,814] | |
| Valeur p (test logarithmique par rangs stratifié) c | 0,0010 | |
| Taux de réponse globale d Répondeurs (RCs+RC+TBRP+RP) n (%) [IC à 95 %] e | 93 (60,4) [0,5220-0,6817] | 54 (35,3) [0,2775-0,4342] |
| Odds ratio par rapport au comparateur [IC exact à 95 %] | 2,795 [1,715-4,562] | |
| Valeur p (test stratifié de Cochran- Mantel-Haenszel) c | < 0,0001 | |
| Réponse complète stringente (RCs) + Réponse complète (RC) n (%) | 7 (4,5) | 3 (2,0) |
| Très bonne réponse partielle (TBRP) n (%) | 42 (27,3) | 10 (6,5) |
| Réponse partielle (RP) n (%) | 44 (28,6) | 41 (26,8) |
| TBRP ou mieux n (%) [IC à 95 %] e | 49 (31,8) [0,2455-0,3980] | 13 (8,5) [0,0460-0,1409] |
| Odds ratio par rapport au comparateur [IC exact à 95 %] | 5,026 [2,514-10,586] | |
| Valeur p (test stratifié de Cochran- Mantel-Haenszel) c | < 0,0001 | |
| Durée de la réponsef * Médiane en mois [IC à 95 %] g | 13,27 [10,612-NA] | 11,07 [8,542-NA] |
b Les patients qui n'ont pas présenté de progression de la maladie ou d'issue fatale avant la date de clôture des données d'analyse ou la date d'instauration d'un autre traitement anti-myélome ultérieur ont été censurés à la date de la dernière évaluation valide de la maladie n'indiquant pas la progression de la maladie avant l'instauration d'un autre traitement anti-myélome (le cas échéant) ou la date de clôture des données d'analyse, selon la première éventualité.
c Stratifié selon l'âge (< 75 ans contre > 75 ans) et le nombre de lignes antérieures de traitement (2 ou 3 contre > 3) selon la théorie de réponse aux items (IRT)
d Les RCs, RC, TBRP et RP ont été évaluées par l'IRC selon les critères de réponse IMWG.
e Estimé en utilisant la méthode de Clopper-Pearson.
f La durée de réponse a été déterminée pour les patients ayant obtenu une réponse ≥ RP (93 patients dans le bras isatuximab et 54 patients dans le bras comparateur). Estimations de Kaplan-Meier de la durée de la réponse.
g Les IC pour les estimations de Kaplan-Meier sont calculés avec une transformation bi logarithmique de la fonction de survie et les méthodes de Brookmeyer et Crowley.
* Date de clôture des données : 11 octobre 2018. Durée médiane de suivi = 11,60 mois. Le HR < 1 est en faveur du bras de traitement par Isa-Pd.
NA : non atteint
Chez
les patients présentant de hauts risques cytogénétiques (évaluation du
laboratoire central), la SSP médiane était de 7,49 (IC à 95 % : 2,628 à
NC) dans le groupe Isa-Pd et de 3,745 (IC à 95 % : 2,793 à 7,885) dans
le groupe Pd (HR = 0,655 ; IC à 95 % : 0,334 à 1,283). Des
améliorations de la SSP ont aussi été observées dans le groupe Isa-Pd
chez les patients ≥ 75 ans (HR = 0,479 ; IC à 95 % : 0,242 à 0,946),
avec un stade ISS III à l'entrée dans l'étude (HR = 0,635 ; IC à 95 % :
0,363 à 1,110), avec une clairance de la créatinine de référence <
60 ml/min/1,73 m² (HR = 0,502 ; IC à 95 % : 0,297 à 0,847), avec > 3
lignes antérieures de traitements (HR = 0,590 ; IC à 95 % : 0,356 à
0,977), chez les patients réfractaires à un traitement antérieur par
lénalidomide (HR = 0,593 ; IC à 95 % : 0,431 à 0,816) ou par inhibiteur
du protéasome (HR = 0,578 ; IC à 95 % : 0,405 à 0,824) et chez ceux
réfractaires au lénalidomide à leur dernière ligne de traitement avant
d'intégrer l'étude (HR = 0,601 ; IC à 95 % : 0,436 à 0,828).
Les données disponibles sont insuffisantes pour conclure sur
l'efficacité du régime Isa-Pd chez les patients traités par daratumumab
(1 patient dans le bras isatuximab et aucun patient dans le bras
comparateur).
Le délai médian jusqu'à la première réponse chez les répondeurs était de 35 jours dans le groupe Isa-Pd contre 58 jours dans le groupe Pd. Après un suivi d'une durée médiane de 11,56 mois dans le groupe Isa-Pd et de 11,73 mois dans le groupe Pd, la survie globale médiane n'a été atteinte dans aucun des groupes de traitement. Le hazard ratio pour la SG était de 0,687 (IC à 95 % : 0,461-1,023, valeur p = 0,0631).
Figure 1 : Courbes de Kaplan-Meier de la SSP - Population ITT - Étude ICARIA-MM (évaluation par l'IRC)
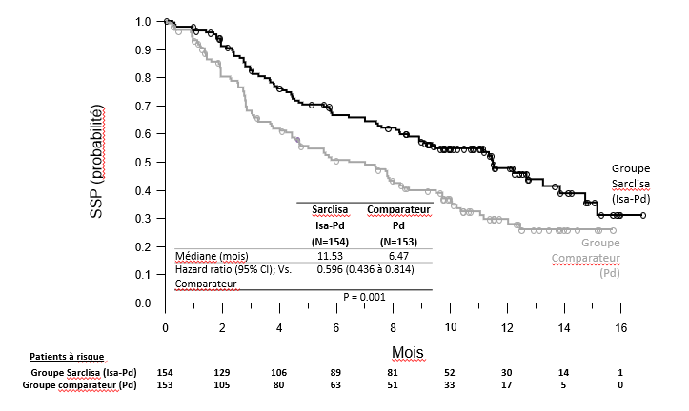
Figure 2 : Courbes de Kaplan-Meier de la SG - Population ITT- Étude ICARIA-MM
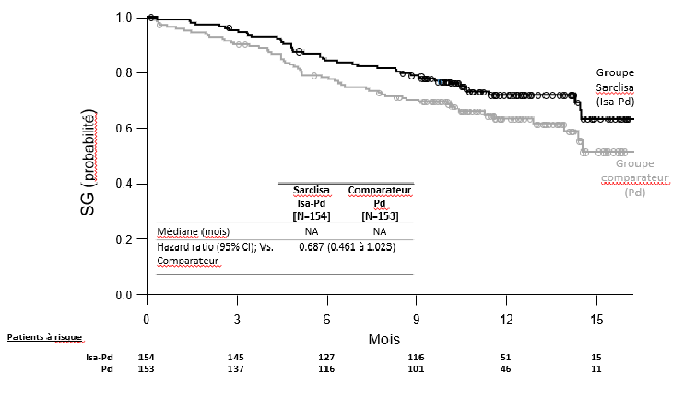
Date de clôture des données : 11 octobre 2018
Dans l'étude ICARIA-MM (EFC14335), un volume basé sur le poids a été utilisé pour la perfusion d'isatuximab. La méthode de perfusion de volume fixe décrite dans la rubrique Posologie et mode d'administration a été évaluée dans la Partie B de l'étude TCD14079, et les simulations pharmacocinétiques ont confirmé des différences minimales dans la pharmacocinétique après l'injection entre un volume basé sur le poids du patient et un volume fixe de 250 ml (voir rubrique Propriétés pharmacocinétiques). Dans la Partie B de l'étude TCD14079, aucun nouveau signal de sécurité et aucune différence au niveau de l'efficacité et de la sécurité d'emploi n'ont été observés par rapport à l'étude ICARIA-MM.
IKEMA (EFC15246)
L'efficacité et la sécurité d'emploi de SARCLISA en association avec le carfilzomib et la dexaméthasone ont été évaluées dans l'étude IKEMA (EFC15246), une étude de phase III multicentrique, internationale, randomisée, en ouvert, avec 2 bras, menée chez des patients atteints de myélome multiple en rechute et/ou réfractaire. Les patients avaient reçu un à trois traitements antérieurs. Les patients atteints d'une maladie réfractaire primaire, qui avaient déjà été traités par carfilzomib ou qui étaient réfractaires à un traitement antérieur par anticorps monoclonaux anti-CD38 étaient exclus.
Un total de 302 patients ont été randomisés selon un rapport de 3:2 pour recevoir soit SARCLISA en association avec du carfilzomib et de la dexaméthasone (groupe Isa-Kd, 179 patients), soit du carfilzomib et de la dexaméthasone (groupe Kd, 123 patients). Le traitement a été administré dans les deux groupes par cycles de 28 jours jusqu'à progression de la maladie ou toxicité inacceptable.
SARCLISA 10 mg/kg a été administré par perfusion IV chaque semaine au cours du premier cycle puis toutes les deux semaines par la suite. Le carfilzomib a été administré par perfusion IV à la dose de 20 mg/m² les jours 1 et 2 et de 56 mg/m² les jours 8, 9, 15 et 16 du cycle 1 ; et à la dose de 56 mg/m² les jours 1, 2, 8, 9, 15 et 16 au cours des cycles suivants de 28 jours. La dexaméthasone (par voie IV les jours de perfusion d'isatuximab et/ou de carfilzomib, et par voie orale les autres jours) 20 mg a été administrée les jours 1, 2, 8, 9, 15, 16, 22 et 23 de chaque cycle de 28 jours.
Dans l'ensemble, à l'inclusion, les caractéristiques démographiques et celles de la maladie étaient similaires entre les deux groupes de traitement. L'âge médian des patients était de 64 ans (intervalle de 33 à 90 ans) ; 8,9 % des patients étaient âgés de ≥ 75 ans. L'indice de performance ECOG était de 0 chez 53,1 % des patients dans le groupe Isa-Kd et 59,3 % dans le groupe Kd, de 1 chez 40,8 % des patients dans le groupe Isa-Kd et 36,6 % dans le groupe Kd, de 2 chez 5,6 % des patients dans le groupe Isa-Kd et 4,1 % dans le groupe Kd, et de 3 chez 0,6 % des patients dans le groupe Isa-Kd et 0 % dans le groupe Kd. La proportion de patients présentant une atteinte de la fonction rénale (débit de filtration glomérulaire < 60 ml/min/1,73 m2) était de 24,0 % dans le groupe Isa-Kd contre 14,6 % dans le groupe Kd. Selon le système de classification international (International Staging System, ISS), le stade de la maladie à l'entrée dans l'étude était de I chez 53,0 %, de II chez 31,1 % et de III chez 15,2 % des patients. Le stade de la maladie à l'entrée dans l'étude selon le système ISS révisé (Revised-ISS, R-ISS) était de I chez 25,8 %, de II chez 59,6 % et de III chez 7,9 % des patients. Dans l'ensemble, 24,2 % des patients présentaient des anomalies chromosomiques à haut risque à l'entrée dans l'étude ; des del(17p), t(4;14) et t(14;16) étaient présents chez 11,3 %, 13,9 % et 2,0 % des patients, respectivement. En outre, l'acquisition (1q21) était présente chez 42,1 % des patients.
Le nombre médian de lignes antérieures de traitement était de 2 (intervalle de 1 à 4), 44,4 % des patients ayant reçu 1 ligne antérieure de traitement. Dans l'ensemble, 89,7 % des patients avaient précédemment reçu des inhibiteurs du protéasome, 78,1 % avaient précédemment reçu des immunomodulateurs (dont 43,4 % ayant reçu au préalable du lénalidomide) et 61,3 % avaient fait l'objet au préalable d'une greffe de cellules souches. Dans l'ensemble, 33,1 % des patients étaient réfractaires aux inhibiteurs du protéasome précédemment reçus, 45,0 % étaient réfractaires aux immunomodulateurs précédemment reçus (dont 32,8 % réfractaires au lénalidomide) et 20,5 % étaient réfractaires à la fois à un inhibiteur du protéasome et un immunomodulateur.
La durée médiane de traitement était de 80,0 semaines pour le groupe Isa-Kd contre 61,4 semaines pour le groupe Kd.
La survie sans progression (SSP) était le critère d'évaluation principal de l'efficacité de l'étude IKEMA. L'amélioration de la SSP représentait une diminution de 46,9 % du risque de progression de la maladie ou de décès chez les patients traités par Isa-Kd par rapport aux patients traités par Kd.
Les résultats en matière d'efficacité sont présentés dans le tableau 8 et les courbes de Kaplan-Meier pour la SSP sont représentées à la Figures 3 :
Tableau 8 : Efficacité de SARCLISA en association avec le carfilzomib et la dexaméthasone par rapport à l'association carfilzomib et dexaméthasone dans le traitement du myélome multiple (analyse de la population en intention de traiter)
| Critère d'évaluation | SARCLISA + carfilzomib + dexaméthasone N = 179 | Carfilzomib + dexaméthasone N = 123 |
| Survie sans progressiona Médiane (mois) [IC à 95 %] | NA [NA-NA] | 19,15 [15,77-NA] |
| Hazard Ratiob [IC à 95 %] | 0,531 [0,318-0,889] | |
| Valeur p (test logarithmique par rangs stratifié)b | 0,0013 | |
| Taux de réponse globalec Répondeurs (RCs + RC + TBRP + RP) [IC à 95 %]d | 86,6 % [0,8071-0,9122] | 82,9 % [0,7509-0,8911] |
| Valeur p (test stratifié de Cochran- Mantel-Haenszel)b | 0,3859 | |
| Réponse complète (RC) | 39,7 % | 27,6 % |
| Très bonne réponse partielle (TBRP) | 33,0% | 28,5 % |
| Critère d'évaluation | SARCLISA + carfilzomib + dexaméthasone N = 179 | Carfilzomib + dexaméthasone N = 123 |
| Réponse partielle (RP) | 14,0 % | 26,8 % |
| TBRP ou mieux (RCs + RC + TBRP) | 72,6 % | 56,1 % |
| [IC à 95 %]d | [0,6547-0,7901] | [0,4687-0,6503] |
| Valeur p (test stratifié de Cochran-Mantel-Haenszel)b e | 0,0021 | |
| RCf | 39,7 % | 27,6 % |
| [IC à 95 %]d | [0,3244-0,4723] | [0,1996 à 0,3643] |
| Taux négatif de maladie résiduelle minimaleg | 29,6 % | 13,0 % |
| [IC à 95 %]d | [0,2303-0,3688] | [0,0762-0,2026] |
| Valeur p (test stratifié de Cochran-Mantel-Haenszel)be | 0,0008 | |
| Durée de la réponseh * (RP ou mieux) Médiane en mois [IC à 95 %]i Hazard Ratiob [IC à 95 %] | NA [NA-NA] | NA [14,752-NA] |
| 0,425 [0,269-0,672] | ||
b Stratifié selon le nombre de lignes antérieures de traitement (1 versus > 1) et le stade R-ISS (I ou II versus III versus non classifié) selon la théorie IRT.
c Les RCs, RC, TBRP et RP ont été évaluées par l'IRC selon les critères de réponse IMWG.
d Estimé en utilisant la méthode de Clopper-Pearson.
e Valeur p nominale.
f RC à tester lors de l'analyse finale.
g Sur la base d'un niveau de sensibilité de 10-5 par séquençage de nouvelle génération (NGS) dans la population ITT.
h Sur la base des répondeurs dans la population ITT. Estimations de Kaplan-Meier de la durée de la réponse. i Les IC pour les estimations de Kaplan-Meier sont calculés avec une transformation bilogarithmique de la fonction de survie et les méthodes de Brookmeyer et Crowley.
* Date de clôture des données : 7 février 2020. Durée médiane de suivi = 20,73 mois. Le HR < 1 est en faveur du bras Isa-Kd.
NA : non atteint.
Des améliorations de la SSP dans le groupe Isa-Kd ont été observées chez les patients présentant de hauts risques cytogénétiques (évaluation du laboratoire central, HR = 0,724 ; IC à 95 % : 0,361 à 1,451), présentant une anomalie chromosomique se traduisant par une acquisition (1q21)(HR = 0,569 ; IC à 95 % : 0,330 à 0,981), ayant ≥ 65 ans (HR = 0,429 ; IC à 95 % : 0,248 à 0,742), avec un débit de filtration glomérulaire de référence (MDRD) < 60 ml/min/1,73 m² (HR = 0,273 ; IC à 95 % : 0,113 à 0,660), avec > 1 ligne de traitement antérieure (HR = 0,479 ; IC à 95 % : 0,294 à 0,778), avec un stade ISS III à l'entrée dans l'étude (HR = 0,650 ; IC à 95 % : 0,295 à 1,434), et chez les patients réfractaires à un traitement antérieur par lénalidomide (HR = 0,598 ; IC à 95 % : 0,339 à 1,055).
Dans l'analyse de sensibilité réalisée sans censurer d'autre traitement contre le myélome, la SSP médiane n'a pas été atteinte (NA) dans le groupe Isa-Kd, contre 19,0 mois (IC à 95 % : 15,38 à NA) dans le groupe Kd (HR = 0,572 ; IC à 99 % : 0,354 à 0,925, p = 0,0025).
Les données disponibles sont insuffisantes pour conclure sur l'efficacité du traitement par Isa-Kd chez les patients traités au préalable par daratumumab (1 patient dans le bras isatuximab et aucun patient dans le bras comparateur).
Le délai médian jusqu'à la première réponse était de 1,08 mois dans le groupe Isa-Kd et de 1,12 mois dans le groupe Kd. Après un suivi d'une durée médiane de 20,73 mois, 17,3 % des patients dans le bras Isa-Kd et 20,3 % des patients dans le bras Kd étaient décédés.
Figure 3 : Courbes de Kaplan-Meier de la SSP - Population ITT - Étude IKEMA (évaluation par l'IRC)
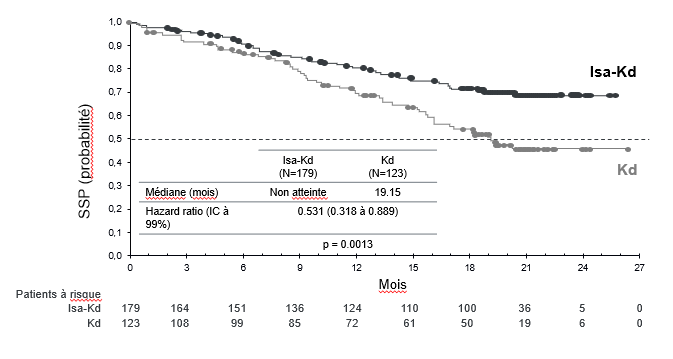
Date de clôture des données = 7 février 2020.
Parmi les patients présentant un débit de filtration glomérulaire (MDRD) de référence
< 50 ml/min/1,73 m2, une réponse rénale complète (≥ 60 ml/min/1,73 m2 lors de ≥ 1 évaluation post- référence) a été observée chez 52,0 % (13/25) des patients dans le groupe Isa-Kd et 30,8 % (4/13) dans le groupe Kd. Une réponse rénale complète soutenue (≥ 60 jours) est survenue chez 32,0 % (8/25) des patients dans le groupe Isa-Kd et 7,7 % (1/13) dans le groupe Kd. Chez les 4 patients du groupe Isa- Kd et les 3 patients du groupe Kd présentant une insuffisance rénale sévère à l'état de référence (débit de filtration glomérulaire (MDRD) > 15 à < 30 ml/min/1,73m 2), une réponse rénale minimale (≥ 30 à < 60 ml/min/1,73 m2 lors de ≥ 1 évaluation post-référence) a été observée chez 100 % des patients dans le groupe Isa-Kd et 33,3 % des patients dans le groupe Kd.
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec SACRLISA dans un ou plusieurs des sous-groupes de la population pédiatrique dans le traitement des tumeurs malignes des tissus hématopoïétiques et lymphoïdes. Voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique.
La pharmacocinétique d'isatuximab a été évaluée chez 476 patients atteints de myélome multiple et traités par perfusion intraveineuse d'isatuximab en monothérapie ou en association avec du pomalidomide/dexaméthasone à des doses allant de 1 à 20 mg/kg, administrés soit une fois par semaine ; toutes les 2 semaines, ou toutes les 2 semaines pendant 8 semaines puis toutes les 4 semaines ; ou toutes les semaines pendant 4 semaines puis toutes les 2 semaines.
L'isatuximab affiche une pharmacocinétique non linéaire avec une disponibilité du médicament médiée par la cible due à sa fixation au récepteur du CD38.
L'exposition à isatuximab (Aire Sous la Courbe -ASC- de concentration plasmatique-temps sur l'intervalle de dose) augmente plus que de manière proportionnelle à la dose proportionnelle pour des doses allant de 1 à 20 mg/kg toutes les 2 semaines, alors qu'aucun écart à la proportionnalité selon la dose n'est observé à des doses allant de 5 à 20 mg/kg selon un schéma d'injection toutes les semaines pendant 4 semaines, puis toutes les 2 semaines. Cela est dû à la forte contribution de la clairance non linéaire médiée par la cible par rapport à la clairance totale à des doses inférieures à 5 mg/kg, qui devient négligeable à des doses plus élevées. Après l'administration d'isatuximab 10 mg/kg toutes les semaines pendant 4 semaines, puis toutes les 2 semaines, le délai médian pour atteindre un état d'équilibre était de 18 semaines avec une accumulation multipliée par 3,1. Dans l'étude ICARIA-MM, un essai clinique mené chez des patients atteints de myélome multiple en rechute et/ou réfractaire et traités par isatuximab en association avec le pomalidomide et la dexaméthasone, la concentration plasmatique maximale moyenne prévue (CV %) et l'ASC à l'état d'équilibre étaient respectivement de 351 µg/ml (36,0 %) et de 72,600 µg.h/ml (51,7 %). Bien que le changement d'une méthode d'administration d'un volume basé sur le poids pour la perfusion d'isatuximab à une méthode de perfusion d'un volume fixe a conduit à des changements de tmax, le changement a eu un impact limité sur la pharmacocinétique d'exposition avec une Cmax à l'état d'équilibre (283 µg/ml contre 284 µg/ml) et une Ctrough à 4 semaines (119 µg/ml contre 119 µg/ml) simulées comparables, pour un patient de poids médian (76 kg). Pour les patients de groupe de poids différents, les Cmax et Cthrough étaient comparables. Dans l'étude IKEMA, un essai clinique mené chez des patients atteints de myélome multiple en rechute et/ou réfractaire et traités par isatuximab en association avec le carfilzomib et la dexaméthasone, la concentration plasmatique maximale moyenne prévue (CV %) et l'ASC à l'état d'équilibre étaient respectivement de 655 µg/ml (30,8 %) et de 159,000 µg.h/ml (37,1 %).
L'administration concomitante d'isatuximab et du pomalidomide, ou d'isatuximab et du carfilzomib, n'a pas eu d'influence sur leur pharmacocinétique.
Distribution
Le volume de distribution total estimé d'isatuximab est de 8,75 l.
Métabolisme
En tant que protéine de grande taille, isatuximab devrait être métabolisé par un processus de catabolisme protéolytique non saturable.
Élimination
L'isatuximab est éliminé par le biais de deux voies d'élimination parallèles, une voie non linéaire médiée par la cible prédominante à de faibles concentrations, et une voie linéaire non spécifique prédominante à des concentrations plus élevées. Dans l'intervalle des concentrations plasmatiques thérapeutiques, la voie linéaire est la principale voie d'élimination et diminue dans le temps de 50 % jusqu'à la valeur à l'état d'équilibre de 9,55 ml/heure (0,229 l/jour). Ceci est associé à une demi-vie terminale de 28 jours.
Populations particulières
Âge
Les analyses pharmacocinétiques de population de 476 patients âgés de 36 à 85 ans ont montré une exposition comparable à isatuximab chez les patients < 75 ans (n=406) et chez ceux ≥ 75 ans (n = 70).
Sexe
L'analyse pharmacocinétique de population de 207 femmes (43,5 %) et de 269 hommes (56,5 %) n'a montré aucun effet cliniquement significatif du sexe sur la pharmacocinétique de isatuximab.
Origine ethnique
L'analyse pharmacocinétique de population de 377 patients de type caucasien (79 %), 25 patients de type asiatique (5 %), 18 patients de race noire (4 %), et 33 patients d'autres origines ethniques (7 %) n'a montré aucun effet cliniquement significatif de l'origine ethnique sur la pharmacocinétique de isatuximab.
Poids
Selon une analyse pharmacocinétique de population utilisant les données de 476 patients, la clairance d'isatuximab augmente avec le poids croissant des patients, justifiant la posologie se basant sur le poids corporel.
Insuffisance hépatique
Aucune étude officielle n'a été menée sur isatuximab chez les patients atteints d'insuffisance hépatique. Sur les 476 patients pris en compte pour les analyses pharmacocinétiques de population, 65 d'entre eux ont présenté une insuffisance hépatique légère [bilirubine totale > 1 à 1,5 fois la limite supérieure de la normale (LSN) ou aspartate aminotransférase (ASAT) > LSN] et 1 patient a présenté une insuffisance hépatique modérée (bilirubine totale > 1,5 à 3 fois la LSN et ASAT à n'importe quel taux). Une insuffisance hépatique légère n'a pas eu d'effets cliniquement significatifs sur la pharmacocinétique d'isatuximab. Les effets d'une insuffisance hépatique modérée (bilirubine totale > 1,5 à 3 fois la LSN et ASAT à n'importe quel taux) ou grave (bilirubine totale > 3 fois la LSN et ASAT à n'importe quel taux) sur la pharmacocinétique d'isatuximab sont inconnus. Cependant, étant donné qu'isatuximab est un anticorps monoclonal, il n'est pas prévu qu'il soit éliminé par le métabolisme hépatique médié par les enzymes hépatiques, et en tant que tel, la variation de la fonction hépatique n'est pas censée affecter l'élimination d'isatuximab (voir rubrique Posologie et mode d'administration).
Insuffisance rénale
Aucune étude officielle n'a été menée sur isatuximab chez les patients présentant une insuffisance rénale. Les analyses pharmacocinétiques de population menées sur 476 patients comprenaient 192 patients présentant une légère insuffisance rénale (60 ml/min/1,73m 2 ≤ débit de filtration glomérulaire estimé [DFG-e] < 90 ml/min/1,73 m2), 163 patients présentant une insuffisance rénale modérée (30 ml/min/1,73 m2 ≤ DFG-e < 60 ml/min/1,73 m2) et 12 patients présentant une insuffisance rénale sévère (DFG-e < 30 ml/min/1,73 m2). Les analyses n'ont suggéré aucun effet cliniquement significatif d'une insuffisance rénale légère à sévère sur la pharmacocinétique d'isatuximab par rapport à une fonction rénale normale.
Population pédiatrique
L'isatuximab n'a pas été évalué chez les patients de moins de 18 ans.
SARCLISA n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Les données non cliniques issues des études conventionnelles de toxicologie en administration répétée n'ont pas révélé de risque particulier pour l'homme, sachant toutefois que les espèces sélectionnées n'étaient pas pharmacologiquement actives et la pertinence pour l'homme n'est pas connue. Aucune étude sur la génotoxicité, la cancérogenèse, et la toxicité pour les fonctions de reproduction et de développement n'a été réalisée.
Préparation pour l'administration intraveineuse
La préparation de la solution de perfusion doit être effectuée dans des conditions aseptiques.- La dose (mg) de solution à diluer de SARCLISA doit être calculée selon le poids du patient (mesuré avant chaque cycle pour permettre l'ajustement en conséquence de la dose à administrer, voir rubrique Posologie et mode d'administration). Plusieurs flacons pourront être nécessaires pour obtenir la dose requise pour le patient.
- Les flacons de solution à diluer de SARCLISA doivent être inspectés visuellement avant dilution pour s'assurer que la solution ne contient pas de particules et qu'elle ne présente pas de coloration anormale.
- Ne pas secouer les flacons.
- Le volume adéquat de SARCLISA doit être prélevé du flacon de SARCLISA et dilué dans la poche de perfusion de 250 ml de chlorure de sodium à 9 mg/ml (0,9 %) ou de solution de glucose à 5 %.
- Le volume adéquat de SARCLISA doit être prélevé et dilué dans la poche de perfusion de 250 ml de chlorure de sodium à 9 mg/ml (0,9 %) ou de solution de glucose à 5 %.
- La poche de perfusion doit être fabriquée à base de polyoléfines (PO), de polyéthylène (PE), de polypropylène (PP) ou de chlorure de polyvinyle (PVC) avec du phtalate de bis(2-éthylhexyle) (DEHP) ou de l'éthyl-vinyle-acétate (EVA).
- Homogénéiser doucement la solution diluée en retournant la poche. Ne pas secouer.
Administration
- La solution pour perfusion doit être administrée par perfusion intraveineuse au moyen d'un kit de perfusion avec tubule IV (en PE ou PVC avec ou sans DEHP, polybutadiène [PBD] ou polyuréthane [PU]) avec un filtre en ligne (en polyéthersulfone [PES], polysulfone ou nylon).
- La solution de perfusion doit être administrée pendant une durée qui dépendra du débit de perfusion (voir rubrique Posologie et mode d'administration).
- La poche de perfusion ainsi préparée ne nécessite aucune protection contre la lumière dans un environnement lumineux artificiel standard.
- Ne pas perfuser la solution de SARCLISA en même temps que d'autres agents dans la même tubulure intraveineuse.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Médicament nécessitant une surveillance particulière pendant le traitement.
Prescription réservée aux médecins compétents en maladie du sang.
Prescription réservée aux spécialistes et services CANCEROLOGIE.
Prescription réservée aux spécialistes et services HEMATOLOGIE.
Prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE.
Réservé à l'usage HOSPITALIER.
Médicament nécessitant une surveillance particulière pendant le traitement.
Prescription réservée aux médecins compétents en maladie du sang.
Prescription réservée aux spécialistes et services CANCEROLOGIE.
Prescription réservée aux spécialistes et services HEMATOLOGIE.
Prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE.
Réservé à l'usage HOSPITALIER.
Solution à diluer pour perfusion.
Solution incolore à légèrement jaune, pratiquement exempte de particules visibles.
25 ml de solution à diluer contenant 500 mg d'isatuximab dans un flacon en verre transparent incolore de type I de 30 ml fermé par un bouchon de bromobutyle revêtu d'ETFE (copolymère à base d'éthylène et de tétrafluoroéthylène). Les flacons sont sertis avec une capsule en aluminium et une bride amovible bleue. Le volume de remplissage du flacon a été établi afin d'assurer l'extraction de 25 ml (c.à.d 26 ml). Emballage contenant un flacon.
1 ml de solution à diluer pour perfusion contient 20 mg d'isatuximab.
Chaque flacon contient 500 mg d'isatuximab dans 25 ml de solution à diluer (500 mg/25 ml).
L'isatuximab est un anticorps monoclonal (AcM) de type immunoglobuline G1(IgG1) produit dans une lignée de cellules de mammifères (ovaire de hamster chinois, [Chinese Hamster Ovary] CHO).
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Saccharose
Chlorhydrate d'histidine monohydraté
Histidine
Polysorbate 80
Eau pour préparations injectables