
Kadcyla 160 mg poudre pour solution à diluer pour perfusion, boîte de 1 flacon de 160 mg
Retiré du marché le : 30/03/2020
Dernière révision : 20/08/2019
Taux de TVA : 0%
Laboratoire exploitant : ROCHE
Source :

Kadcyla, en monothérapie, est indiqué dans le traitement adjuvant de patients adultes atteints d'un cancer du sein précoce HER2 positif qui présentent une maladie résiduelle invasive, au niveau du sein et/ou des ganglions lymphatiques, après un traitement néoadjuvant à base detrastuzumab.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Composition.
Afin d'améliorer la traçabilité des médicaments biologiques, le nom de spécialité et le numéro du lot du produit administré doivent être clairement inscrits dans le dossier dupatient.
Afin d'éviter des erreurs médicamenteuses, il est important de vérifier les étiquettes du flacon afin de s'assurer que le médicament préparé et administré est Kadcyla (trastuzumab emtansine) et non Herceptin (trastuzumab).
Toxicité pulmonaire
Des cas de pneumopathie interstitielle diffuse, incluant des cas de pneumopathie, dont certains conduisant à un syndrome de détresse respiratoire aiguë ou à une issue fatale, ont été rapportés dans les études cliniques avec le trastuzumab emtansine (voir rubrique Effets indésirables). Les signes et les symptômes comprennent dyspnée, toux, fatigue et infiltrats pulmonaires.
Il est recommandé d'arrêter définitivement le traitement avec le trastuzumab emtansine chez les patients présentant une pneumopathie interstitielle ou une pneumopathie, à l'exception d'une pneumopathie radique en situation adjuvante, auquel cas le trastuzumab emtansine doit être arrêté définitivement en cas de grade ³3 ou de grade 2 ne répondant pas au traitement standard (voir rubrique Posologie et mode d'administration).
Les patients ayant une dyspnée de repos en relation avec des complications liées au stade avancé de la maladie, à des facteurs de co-morbidité ainsi que ceux recevant une radiothérapie pulmonaire concomitante, peuvent présenter un risque accru d'évènements indésirables pulmonaires.
Hépatotoxicité
Une hépatotoxicité, principalement sous la forme d'une augmentation asymptomatique des concentrations des transaminases sériques (grade 1- 4), a été observée pendant le traitement avec le trastuzumab emtansine dans les études cliniques (voir rubrique Effets indésirables). Les augmentations des transaminases étaient généralement transitoires avec un pic au jour 8 suivant l'administration du traitement et une amélioration ultérieure à un grade 1 ou inférieur avant le cycle suivant. Un effet cumulatif sur les transaminases a également été observé (la proportion de patients avec des taux d'ASAT/ALAT anormaux de grade 1 - 2 augmente avec les cycles successifs).
Dans la majorité des cas, les patients avec des taux de transaminases élevés ont présenté une amélioration à un grade 1 ou à des valeurs normales dans les 30 jours suivant la dernière dose de trastuzumab emtansine (voir rubrique Effets indésirables).
Des troubles hépatobiliaires graves, incluant des cas d'hyperplasie nodulaire régénérative (HNR) du foie, dont des lésions hépatiques d'origine médicamenteuse avec une issue fatale ont été observés chez des patients traités avec le trastuzumab emtansine. Les cas observés présentaient des facteurs de confusion, sous la forme decomorbidités et/ou de médicaments concomitants ayant un potentiel hépatotoxique connu.
La fonction hépatique doit être surveillée avant l'initiation du traitement et avant chaque administration. Les patients présentant une augmentation du taux d'ALAT initial (par exemple en raison de métastases hépatiques) peuvent être prédisposés à une atteinte hépatique avec un risque plus élevé d'évènement hépatique de grade 3 - 5 ou d'élévation des paramètres hépatiques. Des réductions de dose ou un arrêt du traitement en raison d'une augmentation des transaminases sériques et de la bilirubine totale sont précisées à la rubrique Posologie et mode d'administration.
Des cas d'hyperplasie nodulaire régénérative (HNR) du foie ont été identifiés à partir de biopsies hépatiques chez des patients traités avec le trastuzumab emtansine. L'HNR est une maladie rare du foie caractérisée par une transformation bénigne et étendue du parenchyme hépatique en petits nodules régénératifs. L'HNR peut conduire à une hypertension portale non cirrhotique. Le diagnostic d'HNR peut être uniquement confirmé par histopathologie. Une HNR doit être évoquée chez tous les patients présentant des symptômes cliniques d'hypertension portale et/ou un scanner hépatique compatible avec un tableau de cirrhose, mais avec des taux de transaminases normales et sans autre manifestation de cirrhose. En cas de diagnostic d'HNR, le traitement avec le trastuzumab emtansine doit être définitivement arrêté.
Le trastuzumab emtansine n'a pas été étudié chez les patients ayant des transaminases sériques > 2,5 x la LSN ou une bilirubine totale > 1,5 x la LSN avant l'initiation du traitement. Chez les patients avec des transaminases sériques > 3 x la LSN et une bilirubine totale > 2 x la LSN, le traitement doit être définitivement arrêté. Le traitement des patients insuffisants hépatiques doit être initié avec précaution (voir rubriques Posologie et mode d'administration et Propriétés pharmacocinétiques).
Dysfonctionnement ventriculaire gauche
Les patients traités avec le trastuzumab emtansine présentent un risque accru de développer un dysfonctionnement ventriculaire gauche. Une fraction d'éjection ventriculaire gauche (FEVG) < 40 % a été observée chez des patients traités avec le trastuzumab emtansine et par conséquent, il existe un risque potentiel d'insuffisance cardiaque congestive (ICC) symptomatique (voir rubrique Effets indésirables). Les facteurs de risque généraux d'évènement cardiaque et ceux identifiés dans les études cliniques du cancer du sein en adjuvant avec un traitement par trastuzumab incluent un âge avancé (> 50 ans), des valeurs de FEVG initiales faibles (< 55 %), des valeurs de FEVG faibles avant ou après l'utilisation de paclitaxel en situation adjuvante, un traitement antérieur ou concomitant avec des médicaments antihypertenseurs, un traitement antérieur avec une anthracycline et un indice demasse corporelle élevé (> 25kg/m2).
Un test standard de la fonction cardiaque (échocardiogramme ou scintigraphie cardiaque (MUGA)) doit être réalisé avant l'initiation du traitement et à intervalles réguliers (par exemple tous les trois mois) au cours du traitement. Dans les études cliniques, les patients avaient initialement une FEVG ≥ 50 %. Les patients avec un antécédent d'insuffisance cardiaque congestive (ICC), une arythmie cardiaque grave nécessitant un traitement, un antécédent d'infarctus du myocarde ou d'angor instable au cours des 6 mois précédant la randomisation ou une dyspnée de repos liée au stade avancé de la maladie ont été exclus des études cliniques. L'administration doit être retardée ou le traitement arrêté si nécessaire en cas de dysfonctionnement ventriculaire gauche (voir rubrique Posologie et mode d'administration).
Réactions liées à la perfusion
Le traitement par trastuzumab emtansine n'a pas été étudié chez les patients ayant arrêté définitivement le traitement par trastuzumab en raison de réactions liées à la perfusion. Le traitement n'est pas recommandé chez ces patients. Les patients doivent être étroitement surveillés pour des réactions liées à la perfusion, en particulier au cours de la première perfusion.
Des réactions liées à la perfusion (dues à la libération de cytokines), caractérisées par un ou plusieurs des symptômes suivants, ont été rapportées : bouffées de chaleur, frissons, fièvre, dyspnée, hypotension, râles sibilants, bronchospasme et tachycardie. En général, ces symptômes n'étaient pas sévères (voir rubrique Effets indésirables). Chez la plupart des patients, ces réactions se sont résolues en quelques heures à un jour après la fin de la perfusion. Le traitement doit être interrompu chez les patients présentant des réactions liées à la perfusion sévères jusqu'à ce que les signes et les symptômes soient résolus. La possibilité de reprise du traitement doit être envisagée en fonction de l'évaluation clinique de la sévérité de la réaction. Le traitement doit être définitivement arrêté en cas de réaction liée à la perfusion menaçant le pronostic vital (voir rubrique Posologie et mode d'administration).
Réactions d'hypersensibilité
Le traitement par trastuzumab emtansine n'a pas été étudié chez les patients ayant arrêté définitivement leur traitement par trastuzumab en raison d'une hypersensibilité. Le traitement par trastuzumab emtansine n'est pas recommandé chez ces patients.
Les patients doivent être étroitement surveillés pour des réactions allergiques/d'hypersensibilité qui peuvent présenter le même tableau clinique qu'une réaction liée à la perfusion. Des réactions anaphylactiques graves ont été observées dans les études cliniques avec le trastuzumab emtansine. Les médicaments utilisés pour traiter ces réactions, ainsi qu'un équipement d'urgence, doivent être disponibles pour une utilisation immédiate. En cas de réaction d'hypersensibilité vraie (dont la sévérité augmente avec les perfusions successives), le traitement par trastuzumab emtansine doit être définitivement arrêté.
Hémorragie
Des cas d'évènements hémorragiques incluant des hémorragies du système nerveux central, de l'appareil respiratoire et gastro-intestinales, ont été rapportés pendant le traitement par trastuzumab emtansine. Une issue fatale a été rapportée pour certains de ces événements hémorragiques. Parmi les cas observés, certains patients avaient une thrombocytopénie ou recevaient un traitement anticoagulant ou antiagrégant plaquettaire, d'autres n'avaient aucun facteur de risque supplémentaire connu. Ces produits doivent être utilisés avec précaution et une surveillance supplémentaire doit être envisagée lorsque leur utilisation concomitante est médicalement nécessaire.
Thrombocytopénie
Une thrombocytopénie ou diminution du taux de plaquettes a été fréquemment rapportée avec le trastuzumab emtansine et a été la réaction indésirable la plus fréquente conduisant à un arrêt du traitement (voir rubrique Effets indésirables). Dans les études cliniques, l'incidence et la sévérité de la thrombocytopénie étaient plus élevées chez les patients asiatiques (voir rubrique Effets indésirables).
Il est recommandé de mesurer le taux de plaquettes avant chaque administration de trastuzumab emtansine. Les patients avec une thrombocytopénie (≤ 100 000/mm3) et les patients sous traitement anticoagulant (par exemple la warfarine, l'héparine, les héparines de bas poids moléculaire) doivent être étroitement surveillés pendant le traitement par trastuzumab emtansine. Le trastuzumab emtansine n'a pas été étudié chez les patients avec un taux de plaquettes ≤ 100 000/mm3 avant l'initiation du traitement. En cas de diminution du taux de plaquettes à un grade ≥ 3 (< 50 000/mm3), le trastuzumab emtansine ne doit pas être administré jusqu'à ce que le taux de plaquette revienne à un grade 1 (≥ 75 000/mm3) (voir rubrique Posologie et mode d'administration).
Neurotoxicité
Une neuropathie périphérique, principalement de grade 1 et de type sensoriel, a été rapportéedans les études cliniques avec le trastuzumab emtansine. Les patients présentant initialement une neuropathie périphérique de grade ≥ 3 ont été exclus des études cliniques. Le traitement par trastuzumab emtansine doit être interrompu de façon temporaire chez les patients présentant une neuropathie périphérique de grade 3 ou 4 jusqu'à résolution des symptômes ou amélioration à un grade ≤ 2. Les patients doivent être cliniquement surveillés de façon permanente pour des signes/symptômes deneurotoxicité.
Teneur en sodium desexcipients
Ce médicament contient moins de 1 mmol de sodium (23 mg) par dose, c'est-à-dire qu'il est essentiellement « sans sodium ».
Résumé du profil de sécurité
La sécurité du trastuzumab emtansine a été évaluée chez 2 611 patients atteints d'un cancer du sein dans les études cliniques. Dans cette population de patients :
•les réactions indésirables graves les plus fréquentes (> 0,5 % des patients) étaient : hémorragie, fièvre, thrombocytopénie, dyspnée, douleurs abdominales, douleurs musculosquelettiques et vomissements.
•les réactions indésirables les plus fréquentes (≥ 25 %) avec le trastuzumab emtansine étaient : nausées, fatigue, douleurs musculosquelettiques, hémorragie, céphalées, augmentation des transaminases et thrombocytopénie. La majorité des réactions indésirables rapportées était d'une sévérité de grade 1 ou 2.
•les réactions indésirables de grade ≥3 selon les critères Common Terminology Criteriafor Adverse Events du National Cancer Institute (NCI-CTCAE) les plusfréquentes (> 2 %) étaient : thrombocytopénie, augmentation des transaminases, anémie, neutropénie, fatigue et hypokaliémie.
Tableau des effets indésirables
Les réactions indésirables chez 2 611 patients traités avec le trastuzumab emtansine sont présentées dans le tableau 3. Les réactions indésirables sont listées ci-dessous selon la classification MedDRA des classes de systèmes d'organes et les catégories de fréquence. Les catégories de fréquence sont définies comme suit : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10), peu fréquent (≥ 1/1 000 à < 1/100), rare (≥ 1/10 000 à < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence et de classe de systèmes d'organes, les réactions indésirables sont présentées suivant un ordre décroissant de gravité. Les réactions indésirables ont été rapportées selon les critères NCI-CTCAE pour l'évaluation de latoxicité.
Tableau 3 : Tableau des réactions indésirables chez les patients traités par trastuzumab emtansine dans les études cliniques
| Classe de systèmes d'organes | Très fréquent | Fréquent | Peu fréquent |
| Infections et infestations | Infection urinaire | ||
| Affections hématologiques etdu systèmelymphatique | Thrombocytopénie, anémie | Neutropénie, leucopénie | |
| Affections du système immunitaire | Réaction d'hypersensibilité | ||
| Troubles du métabolisme et de la nutrition | Hypokaliémie | ||
| Affections psychiatriques | Insomnie | ||
| Affections du système nerveux | Neuropathie périphérique, céphalées | Vertiges, dysgueusie, troubles de la mémoire | |
| Affections oculaires | Sécheresse oculaire, conjonctivite, vision floue, augmentation du larmoiement | ||
| Affections cardiaques | Dysfonctionnement ventriculaire gauche | ||
| Affections vasculaires | Hémorragie | Hypertension | |
| Affections respiratoires, thoraciques et médiastinales | Epistaxis, toux, dyspnée | Pneumopathie (pneumopathie interstitielle diffuse) |
| Classe de systèmes d'organes | Très fréquent | Fréquent | Peu fréquent |
| Affections gastro- intestinales | Stomatite, diarrhée, vomissements, nausées, constipation, sécheresse buccale, douleurs abdominales | Dyspepsie, saignement gingival | |
| Affections hépatobiliaires | Augmentation des transaminases | Augmentation des phosphatases alcalines sanguines, augmentation de la bilirubine sanguine | Hépatotoxicité, insuffisance hépatique, hyperplasie nodulaire régénérative, hypertension portale |
| Affections de la peau et du tissu sous- cutané | Rash, prurit, alopécie, trouble unguéal, syndrome d'érythrodysesthésie palmo-plantaire, urticaire | ||
| Affections musculosquelettiques et systémiques | Douleurs musculosquelettiques, arthralgie, myalgie | ||
| Troubles généraux et anomalies au site d'administration | Fatigue, fièvre, asthénie, frissons | Œdème périphérique | Extravasation au site d'injection |
| Lésions, intoxications et complications liées aux procédures | Réactions liées à la perfusion | Pneumopathie radique |
Le tableau 3 présente les données groupées de la période totale de traitement dans les études cliniques dans le cancer du sein métastatique (N = 1 871 ; le nombre médian de cycles de trastuzumab emtansine était de 10) et dans l'étude clinique KATHERINE (N = 740 ; le nombre médian de cycles était de14).
Description de réactions indésirables spécifiques
Augmentation des transaminases (ASAT/ALAT)
Une augmentation des transaminases sériques (grade 1 - 4) a été observée pendant le traitement par trastuzumab emtansine dans les études cliniques (voire rubrique Mises en garde et précautions d'emploi). L'élévation des transaminases était généralement transitoire. Un effet cumulatif du trastuzumab emtansine sur les transaminases a été observé, avec généralement un retour à la normale après l'arrêt du traitement. Des augmentations des transaminases ont été rapportées chez 24,2 % des patients dans les études cliniques dans le cancer du sein métastatique. Des augmentations des taux d'ASAT et d'ALAT de grade 3 ou 4 ont été rapportées chez respectivement 4,2 % et 2,7 % des patients atteints d'un cancer du sein métastatique et sont généralement survenues dans les premiers cycles du traitement (1 - 6). Dans l'étude KATHERINE, des augmentations des transaminases ont été rapportées chez 32,4 % des patients atteints d'un cancer du sein précoce. Des augmentations des transaminases de grade 3 et 4 ont été rapportées chez 1,5 % des patients atteints d'un cancer du sein précoce. En général, la survenue d'événements hépatiques de grade ≥ 3 n'était pas associée à une détérioration de l'état clinique. Les valeurs de suivi ultérieures tendaient à montrer une amélioration à des intervalles autorisant le patient à continuer l'étude et à continuer à recevoir le traitement de l'étude à la même dose ou à une dose réduite. Aucune relation n'a été observée entre l'exposition au trastuzumab emtansine (AUC), la concentration sérique maximale de trastuzumab emtansine (Cmax), l'exposition totale au trastuzumab (AUC) ou la Cmax du DM1 et l'augmentation des transaminases. Pour les modifications de dose en cas d'augmentation des transaminases, voir rubriques Posologie et mode d'administration et Mises en garde et précautions d'emploi.
Dysfonctionnement ventriculaire gauche
Un dysfonctionnement ventriculaire gauche a été rapporté chez 2,2 % des patients dans les études cliniques dans le cancer du sein métastatique avec le trastuzumab emtansine. La majorité des évènements était une diminution de la FEVG de grade 1 ou 2 asymptomatique. Des évènements de grade 3 ou 4 ont été rapportés chez 0,4 % des patients atteints d'un cancer du sein métastatique.
Dans l'étude KATHERINE, un dysfonctionnement ventriculaire gauche est survenu chez 3,0 % des patients atteints d'un cancer du sein précoce, avec un grade 3 ou 4 chez 0,5 % des patients. Une surveillance de la FEVG supplémentaire est recommandée chez les patients avec une FEVG ≤ 45 % (voir tableau 5 à la rubrique Posologie et mode d'administration pour les modifications de dose spécifiques).
Réactions liées à la perfusion
Les réactions liées à la perfusion sont caractérisées par un ou plusieurs des symptômes suivants : bouffées de chaleur, frissons, fièvre, dyspnée, hypotension, râles sibilants, bronchospasme et tachycardie. Des réactions liées à la perfusion ont été rapportées chez 4,0 % des patients dans les études cliniques dans le cancer du sein métastatique avec le trastuzumab emtansine, avec six évènements de grade 3 et aucun évènement de grade 4 rapportés. Dans l'étude KATHERINE, des réactions liées à la perfusion ont été rapportées chez 1,6 % des patients atteints d'un cancer du sein précoce, sans aucun évènement de grade 3 ou 4 rapporté. Les réactions liées à la perfusion se sont résolues en quelques heures à un jour après la fin de la perfusion. Aucun effet de dose n'a été observé dans les études cliniques. Pour les modifications de dose en cas de réactions liées à la perfusion, voir rubriques Posologie et mode d'administration etMises en garde et précautions d'emploi.
Réactions d'hypersensibilité
Une hypersensibilité a été rapportée chez 2,6 % des patients dans les études cliniques dans le cancer du sein métastatique avec le trastuzumab emtansine, avec un évènement de grade 3 et un évènement de grade 4 rapportés. Dans l'étude KATHERINE, une hypersensibilité a été rapportée chez 2,7 % des patients atteints d'un cancer du sein précoce, avec un grade 3 ou 4 chez 0,4 % des patients. Généralement, la majorité des réactions d'hypersensibilité était d'une sévérité légère ou modérée et s'est résolue après traitement. Pour les modifications de dose en cas de réactions d'hypersensibilité, voir rubriques Posologie et mode d'administration etMises en garde et précautions d'emploi.
Hémorragie
L'incidence des évènements hémorragiques sévères (grade ≥ 3) a été de 2,2 % dans la population globale de patients traités par trastuzumab emtansine dans les études cliniques dans le cancer du sein métastatique et de 0,4 % chez les patients atteints d'un cancer du sein précoce, dont un évènement de grade 5. Parmi les cas observés, certains patients avaient une thrombocytopénie ou recevaient un traitement anticoagulant ou antiagrégant plaquettaire, d'autres n'avaient aucun facteude risque supplémentaire connu. Des évènements hémorragiques avec une issue fatale ont été observés.
Thrombocytopénie
Une thrombocytopénie ou diminution du taux de plaquettes a été rapportée chez 24,9 % des patients dans les études cliniques dans le cancer du sein métastatique avec le trastuzumab emtansine et a été la réaction indésirable la plus fréquente conduisant à un arrêt du traitement (2,6 %). Dans l'étude KATHERINE, une thrombocytopénie a été rapportée chez 28,5 % des patients atteints d'un cancer du sein précoce. La majorité des patients a présenté des évènements de grade 1 ou 2 (≥ 50 000/mm3), avec un nadir survenant jusqu'au jour 8 et revenant généralement à un grade 0 ou 1 (≥ 75 000/mm3) avant la dose suivante. Dans les études cliniques, l'incidence et la sévérité de la thrombocytopénie étaient plus élevées chez les patients asiatiques. Indépendamment de la race, l'incidence des évènements de grade 3 ou 4 (< 50 000/mm3) était de 8,7 % chez les patients atteints d'un cancer du sein métastatique traités avec le trastuzumab emtansine et de 18,8 % chez les patients atteints d'un cancer du sein précoce dans l'étude KATHERINE. Pour les modifications de dose en cas de thrombocytopénie, voir rubriques Posologie et mode d'administration et Mises en garde et précautions d'emploi.
Immunogénicité
Comme avec toutes les protéines thérapeutiques, il existe un risque de réponse immunitaire au trastuzumab emtansine. Un total de 1 243 patients issus de sept études cliniques a été testé à plusieurs intervalles de temps pour des anticorps anti-médicament au trastuzumab emtansine. Après administration du trastuzumab emtansine, 5,1 % (63/1 243) des patients ont été testés positifs pour des anticorps anti-trastuzumab emtansine à un ou plusieurs intervalles de temps post-administration. Dans les études de phases I et II, 6,4% (24/376) des patients ont été testés positifs pour des anticorps anti-trastuzumab emtansine. Dans l'étude EMILIA (TDM4370g/BO21977), 5,2 % (24/466) des patients ont été testés positifs pour des anticorps anti-trastuzumab emtansine, dont 13 étaient également positifs pour des anticorps neutralisants. Dans l'étude KATHERINE (BO27938), 3,7 % (15/401) des patients ont été testés positifs pour des anticorps anti-trastuzumab emtansine, dont 5 étaient également positifs pour des anticorps neutralisants. En raison de la faible incidence des anticorps anti-médicament, aucune conclusion ne peut être tirée quant à l'impact des anticorps anti-trastuzumab emtansine sur la pharmacocinétique, la sécurité et l'efficacité du trastuzumab emtansine.
Extravasation
Des réactions secondaires à l'extravasation ont été observées dans les études cliniques avec le trastuzumab emtansine. Ces réactions étaient généralement légères ou modérées et comprenaient un érythème, une sensibilité, une irritation de la peau, une douleur ou un gonflement au site de perfusion. Ces réactions ont été observées plus fréquemment dans les 24 heures suivant la perfusion. Un traitement spécifique de l'extravasation du trastuzumab emtansine n'est pas connu à ce jour.
Anomalies biologiques
Les tableaux 4 et 5 présentent les anomalies biologiques observées chez les patients traités avec le trastuzumab emtansine dans l'étude clinique TDM4370g/BO21977 et l'étude clinique BO27938.
Tableau 4 : Anomalies biologiques observées chez les patients traités avec le trastuzumab emtansine dans l'étude clinique TDM4370g/BO21977
| Paramètre | Trastuzumab emtansine | ||
| Tous grades (%) | Grade 3 (%) | Grade 4 (%) | |
| Hépatique | |||
| Taux de bilirubine augmenté | 21 | < 1 | 0 |
| Taux d'ASAT augmenté | 98 | 8 | < 1 |
| Taux d'ALAT augmenté | 82 | 5 | < 1 |
| Hématologique | |||
| Taux de plaquettes diminué | 85 | 14 | 3 |
| Taux d'hémoglobine diminué | 63 | 5 | 1 |
| Taux de neutrophiles diminué | 41 | 4 | < 1 |
| Potassium | |||
| Taux de potassium diminué | 35 | 3 | < 1 |
Tableau 5 : Anomalies biologiques observées chez les patients traités avec le trastuzumab emtansine dans l'étude clinique BO27938
| Paramètre | Trastuzumab emtansine (N = 740) (3,6 mg/kg) | ||
| Tout grade (%) | Grade 3 (%) | Grade 4 (%) | |
| Hépatique | |||
| Taux de bilirubine augmenté | 11 | 0 | 0 |
| Taux d'ASAT augmenté | 79 | < 1 | 0 |
| Taux d'ALAT augmenté | 55 | < 1 | 0 |
| Hématologique | |||
| Taux de plaquettes diminué | 51 | 4 | 2 |
| Taux d'hémoglobine diminué | 31 | 1 | 0 |
| Taux de neutrophiles diminué | 24 | 1 | 0 |
| Potassium | |||
| Taux de potassium diminué | 26 | 2 | < 1 |
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration:
Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance -Site internet: signalement.social-sante.gouv.fr
SURVEILLANCE du traitement :
- Fonction hépatique : avant l'initiation du traitement et avant chaque administration.
- Fonction cardiaque : faire un test standard (échocardiogramme ou scintigraphie cardiaque) avant l'initiation puis à intervalles réguliers.
- Taux de plaquettes : avant chaque administration.
INSCRIRE dans le dossier du patient le nom de la spécialité et le numéro du lot du produit.
.
PREVENIR IMMEDIATEMENT le médecin en cas de :
- Bleus ou saignements inattendus.
- Picotements, douleur, engourdissement, démangeaisons, sensation que
quelque chose rampe sur ou sous la peau, picotements dans les mains ou
les pieds.
- Bouffées de chaleur, tremblements, fièvre, difficultés à respirer,
diminution de la pression artérielle ou accélération du pouls pendant
la perfusion ou jusqu'à 24 heures après.
- Toux, essouflement au repos, douleur thoracique, chevilles ou bras enflés.
- Jaunissement de la peau ou du blanc des yeux.
- Réactions allergiques : démangeaisons ou sensation d'oppression de la
poitrine, gonflement du visage ou de la langue, difficulté à avaler ou
à respirer.
CONTRACEPTION :
- Les femmes en âge de procréer doivent utiliser une contraception
efficace pendant le traitement et pendant 7 mois après la dernière dose.
- Les patients masculins ou leurs partenaires féminines doivent également utiliser une contraception efficace.
ALLAITEMENT : les femmes peuvent débuter l'allaitement seulement 7 mois après la fin du traitement.
CONDUITE : en cas de réactions liées à la perfusion (bouffées de chaleur, tremblements, fièvre, difficultés à respirer, diminution de la pression artérielle ou accélération du pouls), ne pas conduite ou utiliser de machines jusqu'à disparition des symptômes.
Contraception chez l'homme et la femme
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement par trastuzumab emtansine et pendant 7 mois après la dernière dose de trastuzumab emtansine. Les patients masculins ou leurs partenaires féminines doivent également utiliser une contraception efficace.
Grossesse
Il n'y a pas de données sur l'utilisation du trastuzumab emtansine chez la femme enceinte. Le trastuzumab, un composant du trastuzumab emtansine, peut avoir un effet délétère sur le foetus ou entraîner sa mort lorsqu'il est administré à une femme enceinte. Depuis sa commercialisation, des cas d'oligohydramnios, certains associés à une hypoplasie pulmonaire fatale, ont été rapportés chez des femmes enceintes recevant du trastuzumab. Les études de la maytansine chez l'animal, une entité chimique apparentée au DM1 et de la même classe des maytansinoïdes, suggèrent que le DM1, le composant cytotoxique du trastuzumab emtansine inhibiteur des microtubules, pourrait être tératogène et potentiellement embryotoxique (voir rubriqueDonnées de sécurité précliniques).
L'administration du trastuzumab emtansine aux femmes enceintes n'est pas recommandée et les femmes doivent être informées de la possibilité d'atteinte du foetus avant qu'elles ne soient enceintes. En cas de grossesse, les femmes doivent immédiatement contacter leur médecin. Si une femme enceinte est traitée par trastuzumab emtansine, une surveillance étroite par une équipe multidisciplinaire est recommandée.
Allaitement
Chez la femme, le passage du trastuzumab emtansine dans le lait maternel n'est pas connu. Etant donné que de nombreux médicaments passent dans le lait maternel et en raison du risque de réactions indésirables graves chez les nourrissons allaités, les femmes doivent arrêter l'allaitement avant le début du traitement par trastuzumab emtansine. Les femmes peuvent débuter l'allaitement 7 mois après la fin dutraitement.
Fertilité
Aucune étude toxicologique sur la reproduction et le développement n'a été menée avec le trastuzumab emtansine.
Des études de métabolisme in vitro dans des microsomes hépatiques humains suggèrent que le DM1, un composant du trastuzumab emtansine, est principalement métabolisé par le CYP3A4 et, dans une moindre mesure, par le CYP3A5. L'utilisation concomitante de puissants inhibiteurs du CYP3A4 (par exemple, kétoconazole, itraconazole, clarithromycine, atazanavir, indinavir, néfazodone, nelfinavir, ritonavir, saquinavir, télithromycine et voriconazole) avec le trastuzumab emtansine doit être évitée en raison d'une potentielle augmentation de l'exposition au DM1 et de la toxicité. Il faut envisager un médicament alternatif sans ou avec un potentiel d'inhibition du CYP3A4 minimal. Si l'utilisation concomitante de puissants inhibiteurs du CYP3A4 est inévitable, il faut si possible envisager de retarder le traitement par trastuzumab emtansine jusqu'à ce que les puissants inhibiteurs du CYP3A4 aient été éliminés de la circulation (environ 3 demi-vies d'élimination des inhibiteurs). Si un puissant inhibiteur du CYP3A4 est administré de façon concomitante et que le traitement par trastuzumab emtansine ne peut être retardé, les patients doivent être étroitement surveillés pour des réactions indésirables.
Kadcyla doit être prescrit uniquement par un médecin et administré en perfusion intraveineuse sous la surveillance d'un professionnel de santé expérimenté dans le traitement de patients atteints de cancer (prêt à prendre en charge des réactions allergiques/anaphylactiques liées à la perfusion et dans un environnement où un équipement complet de réanimation est immédiatement disponible (voir rubrique Mises en garde et précautions d'emploi)).
Les patients traités avec le trastuzumab emtansine doivent présenter un statut tumoral HER2 positif, défini par un score 3+ par immunohistochimie (IHC) ou un ratio ≥ 2,0 par hybridation in situ (HIS) ou par hybridation in situ en fluorescence (FISH), déterminé par un dispositif médical de Diagnostic In Vitro (DIV) marqué CE. Si un DIV marqué CE n'est pas disponible, le statut HER2 doit être déterminé par une méthode alternative validée.
Afin d'éviter des erreurs médicamenteuses, il est important de vérifier les étiquettes du flacon afin de s'assurer que le médicament préparé et administré est Kadcyla (trastuzumab emtansine) et non Herceptin (trastuzumab).
Posologie
La dose recommandée de trastuzumab emtansine est de 3,6 mg/kg de poids corporel administrée en perfusion intraveineuse toutes les trois semaines (cycle de 21 jours).
La dose initiale doit être administrée en perfusion intraveineuse de 90 minutes. Les patients doivent être surveillés pendant la perfusion et pendant au moins 90 minutes après la fin de la perfusion initiale pour des symptômes de fièvre, frissons ou d'autres réactions liées à la perfusion. Le site de perfusion doit être étroitement surveillé pour détecter une possible infiltration sous-cutanée pendant l'administration (voir rubrique Effets indésirables).
Si la perfusion précédente a été bien tolérée, les doses suivantes de trastuzumab emtansine peuvent être administrées en perfusions de 30 minutes. Les patients doivent être surveillés pendant la perfusion et pendant au moins 30 minutes après la fin de la perfusion.
La vitesse de perfusion du trastuzumab emtansine doit être diminuée ou la perfusion doit être interrompue si le patient développe des symptômes liés à la perfusion (voir rubriques Mises en garde et précautions d'emploi et Effets indésirables). Le traitement par trastuzumab emtansine doit être arrêté en cas de réactions liées à la perfusion menaçant le pronostic vital.
Durée du traitement
Les patients doivent recevoir le traitement pendant une période totale de 14 cycles, sauf en cas de rechute de la maladie ou de survenue d'une toxicité non contrôlable.
Modification de dose
Le traitement des réactions indésirables symptomatiques peut nécessiter une interruption temporaire du traitement, une réduction de la dose ou l'arrêt du traitement par trastuzumab emtansine conformément aux recommandations présentées ci-dessous et dans les tableaux 1 et 2. La dose de trastuzumab emtansine ne doit pas être ré-augmentée après qu'une réduction de dose ait étéeffectuée.
Tableau 1 : Schéma de réduction de dose
| Schéma de réduction de dose (la dose initiale est de 3,6 mg/kg) | Dose à administrer |
| Première réduction de dose | 3 mg/kg |
| Deuxième réduction de dose | 2,4 mg/kg |
| Nécessité d'une réduction de dose supplémentaire | Arrêt du traitement |
Tableau 2 : Recommandations de modification de dose
| Recommandations de modification de dose dans le cancer du sein précoce | ||
| Effet indésirable | Sévérité | Modification du traitement |
| Augmentation du taux d'alanine aminotransférase (ALAT) | Grade 2-3 (> 3,0 à ≤ 20 ´la LSN le jour de traitement prévu) | Ne pas administrer le trastuzumab emtansine avant que le taux d'ALAT ne revienne à un grade ≤ 1, puis reprendre le traitement en réduisant la dose d'un niveau. |
| Grade 4 (> 20 ´la LSN à tout moment) | Arrêter le traitement avec le trastuzumab emtansine. | |
| Augmentation du taux d'aspartate aminotransférase (ASAT) | Grade 2 (> 3,0 à ≤ 5 ´la LSN le jour de traitement prévu) | Ne pas administrer le trastuzumab emtansine avant que le taux d'ASAT ne revienne à un grade ≤ 1, puis reprendre le traitement au même niveau de dose. |
| Grade 3 (> 5 à ≤ 20 ´la LSN le jour de traitement prévu) | Ne pas administrer le trastuzumab emtansine avant que le taux d'ASAT ne revienne à un grade ≤ 1, puis reprendre le traitement en réduisant la dose d'un niveau. | |
| Grade 4 (> 20 ´la LSN à tout moment) | Arrêter le traitement avec le trastuzumab emtansine. | |
| Hyperbilirubinémie | BILIT > 1,0 à ≤ 2,0 ´la LSN le jour de traitement prévu | Ne pas administrer le trastuzumab emtansine avant que le taux de bilirubine totale ne revienne à ≤ 1,0 × la LSN, puis reprendre le traitement en réduisant la dose d'un niveau. |
| BILIT > 2 ´la LSN à tout moment | Arrêter le traitement avec le trastuzumab emtansine. | |
| Hyperplasie nodulaire régénérative (HNR) | Tous grades | Arrêter définitivement le traitement avec le trastuzumab emtansine. |
| Thrombocytopénie | Grade 2-3 le jourde traitement prévu (25 000 à< 75 000/mm3) | Ne pas administrer le trastuzumab emtansine avant que le taux de plaquettes ne revienne à un grade £1 (≥ 75 000/mm3), puis reprendre le traitement au même niveau de dose. Si un patient nécessite 2 reports de traitement en raison d'une thrombocytopénie, envisager de réduire la dosed'un niveau. |
| Grade 4 à tout moment < 25 000/mm3 | Ne pas administrer le trastuzumab emtansine avant que le taux de plaquettes ne revienne à un grade £1 (≥ 75 000/mm3), puis reprendre le traitement en réduisant la dose d'un niveau. | |
| Dysfonctionnement ventriculaire gauche | FEVG < 45 % | Ne pas administrer le trastuzumab emtansine. Répéter l'évaluation de la FEVG dans les 3 semaines. Si une FEVG < 45 % est confirmée, arrêter le traitement avec le trastuzumab emtansine. |
| FEVG 45 % à < 50 % et diminution ≥ 10 % de la valeur initiale* | Ne pas administrer le trastuzumab emtansine. Répéter l'évaluation de la FEVG dans les 3 semaines. Si la FEVG reste < 50 % et n'est pas revenue à < 10 % de la valeur initiale, arrêter le traitement avec le trastuzumab emtansine. | |
| FEVG 45 % à < 50 % et diminution < 10 % de la valeur initiale* | Continuer le traitement avec le trastuzumab emtansine. Répéter l'évaluation de la FEVG dans les 3 semaines. | |
| FEVG ≥ 50% | Continuer le traitement avec le trastuzumab emtansine. | |
| Insuffisance cardiaque | ICC symptomatique, DSVG de grade 3-4 ou insuffisance cardiaque de grade 3- 4 ou insuffisance cardiaque de grade 2 accompagnée d'une FEVG < 45 % | Arrêter le traitement avec le trastuzumab emtansine. |
| Neuropathie périphérique | Grade 3-4 | Ne pas administrer le trastuzumab emtansine avant amélioration jusqu'à un grade £2. |
| Toxicité pulmonaire | Pneumopathie interstitielle diffuse ou pneumopathie | Arrêter définitivement le traitement avec le trastuzumab emtansine. |
| Pneumopathie radique | Grade 2 | Arrêter le traitement avec le trastuzumab emtansine en l'absence de résolution avec le traitement standard. |
| Grade 3-4 | Arrêter le traitement avec le trastuzumab emtansine. |
ALAT =alanine aminotransférase ; ASAT =aspartate aminotransférase ; ICC = insuffisance cardiaque congestive ; FEVG =fraction d'éjection ventriculaire gauche ; DSVG =dysfonctionnement systolique ventriculaire gauche ; BILIT = bilirubine totale ; LSN =limite supérieure de la normale
* Avant le début du traitement par le trastuzumab emtansine
Oubli ou retard de dose
Si une dose programmée n'est pas administrée, elle doit être administrée dès que possible, sans attendre le prochain cycle prévu. Le calendrier d'administration doit être modifié afin de maintenir un intervalle de 3 semaines entre les doses. La dose suivante doit être administrée conformément aux recommandations de posologie ci-dessus.
Neuropathie périphérique
Le traitement avec le trastuzumab emtansine doit être interrompu de façon temporaire chez les patients présentant une neuropathie périphérique de grade 3 ou 4 jusqu'à amélioration à un grade ≤ 2. Lors de la reprise du traitement, une réduction de dose peut être envisagée selon le schéma de réduction de dose (voir tableau 1).
Populations particulières
Patients âgés
Aucune adaptation de dose n'est requise chez les patients âgés de 65 ans et plus. Les données sont insuffisantes pour établir la sécurité et l'efficacité chez les patients âgés de 75 ans et plus du fait des données limitées dans ce sous-groupe. Une analyse pharmacocinétique de population montre que l'âge n'a pas d'effet cliniquement significatif sur la pharmacocinétique du trastuzumab emtansine (voir rubriques Propriétés pharmacodynamiques et Propriétés pharmacocinétiques).
Insuffisance rénale
Aucune adaptation de la dose initiale n'est requise chez les patients présentant une insuffisance rénale légère ou modérée (voir rubrique Propriétés pharmacocinétiques). Le besoin potentiel d'une adaptation de dose chez les patients présentant une insuffisance rénale sévère ne peut être déterminé en raison de l'insuffisance des données. Par conséquent, les patients avec une insuffisance rénale sévère doivent être étroitementsurveillés.
Insuffisance hépatique
Aucune adaptation de la dose initiale n'est requise chez le patient présentant une insuffisance hépatique légère ou modérée. Le trastuzumab emtansine n'a pas été étudié chez les patients avec une insuffisance hépatique sévère. Le traitement des patients insuffisants hépatiques doit être initié avec précaution en raison de l'hépatotoxicité observée avec le trastuzumab emtansine (voir rubriques Mises en garde et précautions d'emploi et Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l'efficacité chez les enfants et les adolescents âgés de moins de 18 ans n'ont pas été établies. Il n'y a pas d'utilisation justifiée dans la population pédiatrique dans l'indication de cancer du sein métastatique.
Mode d'administration
Kadcyla est à administrer par voie intraveineuse. Le trastuzumab emtansine doit être reconstitué et dilué par un professionnel de santé et administré en perfusion intraveineuse. Il ne doit pas être administré en injection rapide ou bolus intraveineux.
Pour les instructions concernant la reconstitution et la dilution du médicament avant administration, voir la rubrique Instructions pour l'utilisation, la manipulation et l'élimination.
Durée de conservation :
Flacon avant ouverture
3 ans
Solution reconstituée
La stabilité physico-chimique de la solution reconstituée a été démontrée jusqu'à 24 heures entre 2°C et 8°C. Toutefois, d'un point de vue microbiologique, le produit doit être utilisé immédiatement. En cas d'utilisation non immédiate, les flacons reconstitués peuvent être conservés jusqu'à 24 heures entre 2°C et 8°C, à la condition qu'ils aient été reconstitués dans des conditions d'asepsie dûment contrôlées et validées. Ils doivent être ensuite éliminés.
Solution diluée
La solution reconstituée de Kadcyla diluée dans des poches pour perfusion contenant une solution de chlorure de sodium à 9 mg/mL (0,9 %) pour perfusion ou une solution de chlorure de sodium à 4,5 mg/mL(0,45%)pourperfusion,eststablejusqu'à24heuresentre2°Cet8°C,àlaconditionqu'elleait été préparée dans des conditions d'asepsie dûment contrôlées et validées. Des particules peuvent être observées au cours de la conservation en cas de dilution avec du chlorure de sodium à 0,9 % (voir rubriqueInstructions pour l'utilisation, la manipulation et l'élimination).
Précautions particulières de conservation a conserver au réfrigérateur (entre 2°c et 8°c). :Pour les conditions de conservation du médicament après reconstitution et dilution, voir la rubrique Durée de conservation.
Ce médicament ne doit pas être mélangé ou dilué avec d'autres médicaments à l'exception de ceux mentionnés dans la rubrique Instructions pour l'utilisation, la manipulation et l'élimination.
Une solution de glucose (5 %) ne doit pas être utilisée pour la reconstitution ou la dilution car cela entraîne l'agrégation de la protéine.
La solution reconstituée de Kadcyla doit être diluée dans une poche pour perfusion en chlorure de polyvinyle (PVC) ou en polyoléfine sans PVC et sans latex.
L'utilisation d'un filtre en ligne en polyéthersulfone de 0,20 ou 0,22 microns est requise pour la perfusion lorsque la solution à diluer pour perfusion est diluée avec une solution de chlorure de sodium à 9 mg/mL (0,9 %) pour perfusion
La quantité appropriée de solution doit être
prélevée du flacon et ajoutée dans une poche pour perfusion contenant 250 mL
d'une solution de chlorure de sodium à 4,5 mg/mL (0,45 %) pour perfusion ou de
chlorure de sodium à 9 mg/mL (0,9 %) pour perfusion. Une solution de glucose (5
%) ne doit pas être utilisée (voir rubrique Incompatibilités). Une
solution de chlorure de sodium à 4,5 mg/mL (0,45 %) pour perfusion peut être
utilisée sans un filtre en ligne en polyéthersulfone de 0,20 ou 0,22 microns. Si
une solution de chlorure de sodium à 9 mg/mL (0,9 %) pour perfusion est utilisée
pour la perfusion, un filtre en ligne en polyéthersulfone de 0,20 ou 0,22
microns est requis.
Il n'y a pas d'antidote connu en cas de surdosage en trastuzumab emtansine. En cas de surdosage, le patient doit être étroitement surveillé pour les signes ou symptômes de réactions indésirables et un traitement symptomatique approprié doit être instauré. Des cas de surdosage ont été rapportés avec le traitement par trastuzumab emtansine, le plus souvent associés à une thrombocytopénie et il y a eu un décès. Dans le cas fatal, le patient a reçu par erreur le trastuzumab emtansine à 6 mg/kg et est décédé environ 3 semaines après le surdosage. Aucun lien de causalité avec le trastuzumab emtansine n'a étéétabli.
Classe pharmacothérapeutique : Agents antinéoplasiques, autres agents antinéoplasiques, anticorps monoclonaux, code ATC : L01XC14
Mécanisme d'action
Kadcyla, le trastuzumab emtansine, est un anticorps conjugué ciblant le récepteur HER2 qui contient le trastuzumab, un anticorps monoclonal humanisé de classe IgG1anti-HER2, lié de façon covalente au DM1, un inhibiteur de microtubules (dérivé de la maytansine), grâce à l'agent de liaison thioéther stable MCC (4-[N-maleimidométhyl] cyclohexane-1-carboxylate). L'emtansine fait référence au complexe MCC-DM1. En moyenne, 3,5 molécules de DM1 sont conjuguées à chaque molécule de trastuzumab.
La conjugaison du DM1 au trastuzumab confère à l'agent cytotoxique une sélectivité pour les cellules tumorales surexprimant HER2, augmentant ainsi la libération intracellulaire de DM1 directement dans les cellules malignes. Suite à sa liaison à HER2, le trastuzumab emtansine est internalisé par le biais du récepteur, s'en suit une dégradation lysosomale, conduisant à la libération de catabolites cytotoxiques contenant du DM1 (essentiellement le complexe lysine-MCC-DM1).
Le trastuzumab emtansine présente à la fois le mécanisme d'action du trastuzumab et du DM1 :
•Le trastuzumab emtansine, comme le trastuzumab, se fixe au sous-domaine IV du domaine extracellulaire de HER2 (ECD), ainsi qu'aux récepteurs Fcγ et au complément C1q. De plus, le trastuzumab emtansine, comme le trastuzumab, inhibe l'excrétion du domaine extracellulaire de HER2, inhibe la transmission du signal par la voie phosphatidylinositol 3- kinase (PI3-K) et agitcomme médiateur de la cytotoxicité cellulaire anticorps-dépendante (ADCC) dans les cellules cancéreuses du sein humain qui surexprimentHER2.
•Le DM1, le composant cytotoxique du trastuzumab emtansine, se fixe à la tubuline. En inhibant la polymérisation de la tubuline, le DM1 et le trastuzumab emtansine entrainent l'arrêt du cycle cellulaire en phase G2/M, conduisant à terme à la mort cellulaire par apoptose. Les résultats des essais de cytotoxicités in vitro montrent que le DM1 est 20 à 200 fois plus puissant que les taxanes et lesvinca-alcaloïdes.
•L'agent de liaison MCC est conçu pour limiter la libération systémique et augmenter la libération ciblée du DM1, comme démontré par la détection de très faibles concentrationsde DM1 libre dans le plasma.
Efficacité et sécurité clinique
Cancer du sein précoce
Les données d'efficacité et de sécurité cliniques sont en cours d'évaluation par l'Agence Européenne du médicament. Les informations ci-dessous seront sujettes à modification.
BO27938 (KATHERINE)
BO27938 (KATHERINE) est un essai randomisé, multicentrique et en ouvert portant sur 1 486 patients atteints d'un cancer du sein précoce HER2 positif présentant une maladie résiduelle invasive (patients n'ayant pas obtenu une réponse pathologique complète [pathological complete response ou pCR]) au niveau du sein et/ou des ganglions lymphatiques axillaires après un traitement systémique préopératoire ayant comporté une chimiothérapie et un traitement anti-HER2. Les patients devaient avoir terminé leur traitement systémique préopératoire, comprenant une chimiothérapie et un traitement anti-HER2. Les patients pouvaient avoir reçu une anthracycline dans le cadre de leur traitement préopératoire en plus de la chimiothérapie à base de taxane. La totalité de la chimiothérapie systémique devait avoir été administrée en préopératoire. Les patients pouvaient avoir reçu plus d'un traitement anti-HER2. Les patients ont reçu conformément aux recommandations locales une radiothérapie et/ou une hormonothérapie concomitante au traitement étudié. Les échantillons de tumeur du sein devaient montrer une surexpression de HER2 définie par un score 3+ par IHC ou un ratio d'amplification ≥ 2,0 par HIS déterminé dans un laboratoire central. Les patients ont été randomisés (1:1) pour recevoir le trastuzumab ou le trastuzumab emtansine. La randomisation a été stratifiée selon le stade clinique initial, le statut des récepteurs hormonaux, le traitementanti-
HER2 préopératoire (trastuzumab, trastuzumab plus traitement[s] anti-HER2) et le statut ganglionnaire pathologique évalué après le traitement préopératoire.
Le trastuzumab emtansine a été administré par voie intraveineuse à 3,6 mg/kg le jour 1 d'un cycle de 21 jours. Le trastuzumab a été administré par voie intraveineuse à 6 mg/kg le jour 1 d'un cycle de 21 jours. Les patients ont été traités par trastuzumab emtansine ou trastuzumab pendant un total de 14 cycles, sauf en cas de récidive de la maladie, de retrait du consentement ou de toxicité inacceptable, en fonction de l'évènement se produisant en premier. Au moment de l'analyse primaire, la durée médiane du traitement était de 10 mois (intervalle de 1 à 12) pour le trastuzumab emtansine et de 10 mois (intervalle de 1 à 13) pour le trastuzumab. Les patients ayant arrêté le trastuzumab emtansine pouvaient poursuivre le traitement anti-HER2 avec le trastuzumab jusqu'au terme prévu du traitement étudié (jusqu'à 14 cycles), si cela était approprié en fonction de considérations relatives à la toxicité et à la discrétion de l'investigateur.
Le critère primaire d'évaluation d'efficacité de l'étude clinique était la survie sans maladie invasive (Invasive Disease-Free Survival ou IDFS). L'IDFS était définie comme le temps entre la date de randomisation et la date de première apparition d'une récidive ipsilatérale d'une tumeur du sein invasive, d'une récidive locale ou régionale ipsilatérale d'un cancer du sein invasif, d'une récidive à distance, d'un cancer du sein invasif controlatéral ou d'un décès, quelle qu'en soit la cause. Les autres critères d'évaluation comprenaient l'IDFS incluant un deuxième cancer primitif autre qu'un cancer du sein, la survie sans maladie (disease-free survival ou DFS), la survie globale (overall survival ou OS) et l'intervalle sans récidive à distance (distant recurrence-free interval ouDRFI).
Les données démographiques des patients et les caractéristiques tumorales à l'inclusion étaient équilibrées entre les bras de traitement. L'âge médian était d'environ 49 ans (intervalle de 23 à 80 ans), 72,8 % des patients étaient de type Caucasien, 8,7 % étaient Asiatiques et 2,7 % étaient Noirs ou Afro-américains. Il n'y avait que des femmes, à l'exception de 5 patients. 22,5 % des patients ont été inclus en Amérique du Nord, 54,2 % en Europe et 23,3 % dans le reste du monde. Les caractéristiques pronostiques tumorales, notamment le statut des récepteurs hormonaux (positif : 72,3 %, négatif : 27,7 %), le stade clinique initial (inopérable : 25,3 %, opérable : 74,8 %) et le statut pathologique ganglionnaire après le traitement préopératoire (positif : 46,4 %, négatif ou non évalué : 53,6 %) étaient similaires dans les bras de l'étude.
La majorité des patients (76,9 %) avait reçu une chimiothérapie néoadjuvante contenant une anthracycline. 19,5 % des patients avaient reçu un autre traitement anti-HER2 en plus du trastuzumab, dans le cadre du traitement néoadjuvant. Le pertuzumab était le deuxième traitement chez 93,8 % des patients ayant reçu un deuxième traitement anti-HER2 néoadjuvant.
Une amélioration cliniquement et statistiquement significative de l'IDFS a été observée chez les patients ayant reçu le trastuzumab emtansine par rapport au trastuzumab (HR = 0,50, IC à 95 % [0,39 - 0,64], p < 0,0001). Les estimations des taux d'IDFS à 3 ans étaient de 88,3 % vs. 77,0 % dans les bras trastuzumab emtansine vs. trastuzumab, respectivement. Voir le tableau 6 et la figure1.
Tableau 6 : Résumé de l'efficacité de l'étude clinique BO27938 (KATHERINE)
| Trastuzumab N = 743 | Trastuzumab emtansine N = 743 | |
| Critère primaire | ||
| Survie sans maladie invasive (IDFS)3 | ||
| Nombre (%) de patients avec évènement | 165 (22,2 %) | 91 (12,2 %) |
| HR [IC à 95 %] | 0,50 [0,39 ; 0,64] | |
| Valeur de p (test Log-Rank, non stratifié) | < 0,0001 | |
| Taux de patients sans évènement à 3 ans2, % [IC à 95 %] | 77,02 [73,78 ; 80,26] | 88,27 [85,81 ; 90,72] |
| Critères secondaires1 | ||
| Trastuzumab N = 743 | Trastuzumab emtansine N = 743 | |
| IDFS incluant un deuxième cancer primitif autre qu'un cancer du sein | ||
| Nombre (%) de patients avec évènement | 167 (22,5 %) | 95 (12,8 %) |
| HR [IC à 95 %] | 0,51 [0,40 ; 0,66] | |
| Valeur de p (test Log-Rank, non stratifié) | < 0,0001 | |
| Taux de patients sans évènement à 3 ans2, % [IC à 95 %] | 76,9 [73,65 ; 80,14] | 87,7 [85,18 ; 90,18] |
| Survie sans maladie (DFS) | ||
| Nombre (%) de patients avec évènement | 167 (22,5 %) | 98 (13,2 %) |
| HR [IC à 95 %] | 0,53 [0,41 ; 0,68] | |
| Valeur de p (test Log-Rank, non stratifié) | < 0,0001 | |
| Taux de patients sans évènement à 3 ans2, % [IC à 95 %] | 76,9 [73,65 ; 80,14] | 87,41 [84,88 ; 89,93] |
| Survie globale (OS)3 | ||
| Nombre (%) de patients avec évènement | 56 (7,5 %) | 42 (5,7 %) |
| HR [IC à 95 %] | 0,70 [0,47 ; 1,05] | |
| Valeur de p (test Log-Rank, non stratifié) | 0,0848 | |
| Taux de survie à 5 ans2,% [IC à 95 %] | 86,8 [80,95 ; 92,63] | 92,1 [89,44 ; 94,74] |
| Intervalle sans récidive à distance (DRFI) | ||
| Nombre (%) de patients avec évènement | 121 (16,3 %) | 78 (10,5 %) |
| HR [IC à 95 %] | 0,60 [0,45 ; 0,79] | |
| Valeur de p (test Log-Rank, non stratifié) | 0,0003 | |
| Taux de patients sans évènement à 3 ans2, % [IC à 95 %] | 83,0 [80,10 ; 85,92 ] | 89,7 [87,37 ; 92,01 ] |
Signification des abréviations (tableau 6) : HR : Hazard Ratio ; IC : intervalles de confiance
1.Test hiérarchique appliqué pour l'IDFS etl'OS
2.Le taux de patients sans évènement à 3 ans et le taux de survie à 5 ans sont calculés à partir des estimations de Kaplan-Meier
3.Données provenant de la première analyseintermédiaire
Figure 1 : Courbe de Kaplan-Meier de la survie sans maladie invasive dans l'étude cliniqueKATHERINE

Figure 2 : Courbe de Kaplan-Meier de l'intervalle sans récidive à distance dans l'étude cliniqueKATHERINE
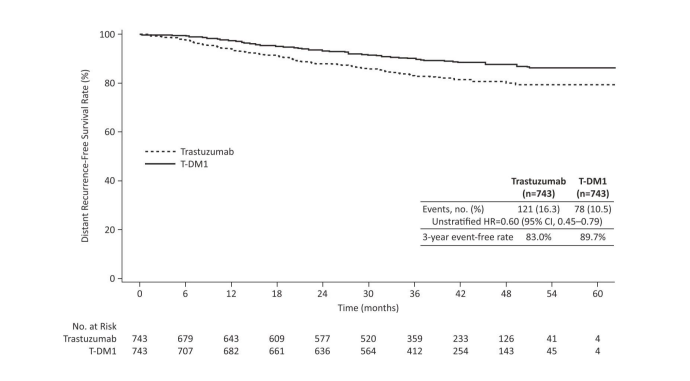
Dans l'étude clinique KATHERINE, un bénéfice homogène du traitement par trastuzumab emtansine pour l'IDFS a été observé dans tous les sous-groupes prédéfinis évalués, corroborant la robustesse du résultat global (voir figure 3).

Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec le trastuzumab emtansine dans tous les sous-groupes de la population pédiatrique dans le cancer du sein (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
L'analyse pharmacocinétique de population n'a suggéré aucune différence au niveau de l'exposition au trastuzumab emtansine en fonction du statut de la maladie (situation adjuvante vs.métastatique).
Absorption
Le trastuzumab emtansine est administré par voie intraveineuse. Aucune étude n'a été réalisée avec d'autres voies d'administration.
Distribution
Les patients de l'étude TDM4370g/BO21977 et de l'étude BO29738 qui ont reçu 3,6 mg/kg de trastuzumab emtansine par voie intraveineuse toutes les 3 semaines avaient une concentration sérique maximale moyenne (Cmax) de trastuzumab emtansine au cycle 1 de 83,4 (± 16,5) µg/mL et de 72,6 (± 24,3) µg/mL, respectivement. Sur la base d'une analyse pharmacocinétique de population, après administration intraveineuse, le volume de distribution central du trastuzumab emtansine était de 3,13 l et approchait celui du volume plasmatique.
Biotransformation (trastuzumab emtansine et DM1)
Le trastuzumab emtansine est supposé subir une déconjugaison et un catabolisme par protéolyse dans les lysosomes cellulaires.
Les études de métabolisme in vitro dans des microsomes hépatiques humains suggèrent que le DM1, une petite molécule composante du trastuzumab emtansine, est principalement métabolisé par le CYP3A4 et dans une moindre mesure par le CYP3A5. Le DM1 n'a pas inhibé les principales enzymes CYP450 in vitro. Dans le plasma humain, les catabolites du trastuzumab emtansine MCC-DM1, Lys- MCC-DM1 et DM1 ont été détectés à de faibles concentrations. In vitro, le DM1 était un substrat de la P-glycoprotéine (P-gp).
Élimination
Sur la base d'une analyse pharmacocinétique de population, après administration intraveineuse de trastuzumab emtansine chez des patients atteints d'un cancer du sein métastatique HER2 positif, la clairance du trastuzumab emtansine était de 0,68 l/jour et la demi-vie d'élimination (t1/2) était d'environ 4 jours. Aucune accumulation de trastuzumab emtansine n'a été observée après administration répétée par perfusion intraveineuse toutes les 3 semaines.
Sur la base d'une analyse pharmacocinétique de population, le poids corporel, l'albumine, la somme du diamètre le plus long des lésions cibles selon les critères RECIST (Response Evaluation Criteria In Solid Tumors), le passage de HER2 dans le domaine extracellulaire (ECD), les concentrations de trastuzumab initiales et l'aspartate aminotransférase (ASAT) ont été identifiés comme des covariables statistiquement significatives des paramètres pharmacocinétiques du trastuzumab emtansine.
Cependant, l'ampleur de l'effet de ces covariables sur l'exposition au trastuzumab emtansine suggère que ces covariables ne devraient pas avoir d'effet cliniquement significatif sur l'exposition au trastuzumab emtansine. De plus, une analyse exploratoire a montré que l'impact de ces covariables (c.-à.-d. fonction rénale, race et âge) sur la pharmacocinétique du trastuzumab total et du DM1 était limité et n'avait pas d'incidence clinique. Dans les études non cliniques, les catabolites du trastuzumab emtansine incluant DM1, Lys-MCC-DM1 et MCC-DM1, sont principalement excrétés dans la bile avec une élimination urinaire minimale.
Linéarité/non-linéarité
Le trastuzumab emtansine, après administration par voie intraveineuse toutes les 3 semaines, a montré une pharmacocinétique linéaire pour des doses allant de 2,4 à 4,8 mg/kg. Les patients ayant reçu des doses inférieures ou égales à 1,2 mg/kg avaient une clairance plus rapide.
Populations particulières
Patients âgés
L'analyse pharmacocinétique de population a montré que l'âge n'affectait pas la pharmacocinétique du trastuzumab emtansine. Aucune différence significative n'a été observée dans la pharmacocinétique du trastuzumab emtansine entre les patients âgés de moins de 65 ans (n = 577), ceux âgés de 65 à 75 ans (n = 78) et ceux âgés de plus de 75 ans (n =16).
Insuffisance rénale
Aucune étude pharmacocinétique formelle n'a été conduite chez les patients avec une insuffisance rénale. L'analyse pharmacocinétique de population a montré que la clairance de la créatinine n'affecte pas la pharmacocinétique du trastuzumab emtansine. La pharmacocinétique chez les patients avec une insuffisance rénale légère (clairance de la créatinine [CLcr] de 60 à 89 mL/min, n = 254) ou modérée (CLcr de 30 à 59 mL/min, n = 53) était similaire à celle chez les patients avec une fonction rénale normale (CLcr ≥ 90 mL/min, n = 361). Les données pharmacocinétiques chez les
patients avec une insuffisance rénale sévère (CLcr de 15 à 29 mL/min) sont limitées (n = 1). Par conséquent, aucune recommandation de dose ne peut être faite.
Insuffisance hépatique
Le foie est l'organe principal pour l'élimination de DM1 et des catabolites contenant DM1. La pharmacocinétique du trastuzumab emtansine et des catabolites contenant DM1 a été évaluée après l'administration de 3,6 mg/kg de trastuzumab emtansine chez des patients avec un cancer du sein métastatique HER2+ et une fonction hépatique normale (n = 10), une insuffisance hépatique légère (child-Pugh A, n = 10) et modérée (Child-Pugh B, n = 8).
•Les concentrations plasmatiques de DM1 et des catabolites contenant DM1 (Lys-MCC DM1 et MCC-DM1) étaient faibles et comparables entre les patients avec ou sans insuffisance hépatique.
•Les expositions systémiques (ASC) au trastuzumab emtansine au Cycle 1 chez les patients avec une insuffisance hépatique légère et modérée étaient respectivement et approximativement de 38 % et 67 % inférieures à celles des patients avec une fonction hépatique normale. L'exposition au trastuzumab emtansine (ASC) au Cycle 3 après administration répétée chez les patients avec une insuffisance hépatique légère oumodérée était dans l'intervalle observé chez des patients avec une fonction hépatiquenormale.
Aucune étude pharmacocinétique formelle n'a été conduite et aucune donnée pharmacocinétique de population n'a été recueillie chez les patients avec une insuffisance hépatique sévère (Child-Pugh de classe C).
Autres populations particulières
L'analyse pharmacocinétique de population a montré que la race ne semblait pas influencer la pharmacocinétique du trastuzumab emtansine. Comme la plupart des patients dans les études cliniques avec le trastuzumab emtansine étaient des femmes, l'effet du genre sur la pharmacocinétique du trastuzumab emtansine n'a pas été évalué de façon formelle.
Le trastuzumab emtansine n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines. L'impact des réactions indésirables rapportées comme la fatigue, les céphalées, les vertiges et une vision floue sur l'aptitude à conduire des véhicules et à utiliser des machines n'est pas connu. Les patients présentant des réactions liées à la perfusion (bouffées de chaleur, tremblements, fièvre, difficultés à respirer, diminution de la pression artérielle ou accélération du pouls) devront être avertis de s'abstenir de conduire des véhicules ou d'utiliser des machines jusqu'à disparition des symptômes.
Toxicologie et/ou pharmacologie chez l'animal
L'administration du trastuzumab emtansine a été bien tolérée chez le rat et le singe à des doses allant respectivement jusqu'à 20 mg/kg et 10 mg/kg, correspondant à 2040 µg de DM1/m2 dans les deux espèces, ce qui est approximativement équivalent à la dose clinique du trastuzumab emtansine chez les patients. Dans les études de toxicité selon les Bonnes Pratiques de Laboratoire, à l'exception d'une toxicité axonale périphérique irréversible (observée seulement chez le singe à une dose ≥ 10 mg/kg) et d'une toxicité sur les organes de la reproduction (observée seulement chez le rat à une dose de 60 mg/kg), des toxicités dose dépendantes partiellement ou complètement réversibles ont été identifiées dans les deux modèles animaux. Les toxicités principales incluaient le foie (augmentation des enzymes hépatiques) à des doses respectives chez le rat et le singe ≥ 20 mg/kg et ≥ 10 mg/kg, la moelle osseuse (diminution du taux de plaquettes et de globules blancs) à des doses ≥ 20 mg/kg et ≥ 10 mg/kg et les organes lymphoïdes à des doses ≥ 20 mg/kg et ≥ 3mg/kg.
Mutagénicité
Le DM1 était aneugène ou clastogène dans un test de micronoyaux in vivo sur la moelle osseuse de rat à dose unique à des expositions comparables aux concentrations maximales moyennes du DM1 mesurées après administration de trastuzumab emtansine chez l'homme. Le DM1 n'était pas mutagène dans un test in vitro de mutation bactérienne inverse (Ames).
Perturbation de la fertilité et tératogénicité
Aucune étude de fertilité n'a été réalisée chez l'animal afin d'évaluer l'effet du trastuzumab emtansine. Cependant, sur la base de résultats des études de toxicité animale générales, des effets délétères sur la fertilité sont attendus.
Aucune étude spécifique sur le développement embryo-foetal n'a été conduite chez l'animal avec le trastuzumab emtansine. Une toxicité du trastuzumab sur le développement a été identifiée en situation clinique bien qu'elle n'ait pas été prédite au cours du programme non-clinique. De plus, une toxicité de la maytansine sur le développement a été identifiée dans des études non-cliniques qui suggèrent que le DM1, le composant maytansinoïde cytotoxique inhibiteur des microtubules du trastuzumab emtansine, sera de façon similaire tératogène et potentiellement embryotoxique.
Une technique d'asepsie appropriée doit être utilisée. Des procédures appropriées pour la préparation des médicaments de chimiothérapie doivent être utilisées.
La solution reconstituée de Kadcyla doit être diluée dans une poche pour perfusion en chlorure de polyvinyle (PVC) ou en polyoléfine sans PVC et sans latex.
L'utilisation d'un filtre en ligne en polyéthersulfone de 0,20 ou 0,22 microns est requise pour la perfusion lorsque la solution à diluer pour perfusion est diluée avec une solution de chlorure de sodium à 9 mg/mL (0,9 %) pour perfusion.
Afin d'éviter des erreurs médicamenteuses, il est important de vérifier les étiquettes du flacon afin de s'assurer que le médicament préparé est Kadcyla (trastuzumab emtansine) et non Herceptin (trastuzumab).
Instructions pour la reconstitution
•En utilisant une seringue stérile, injecter lentement 8 mL d'eau pour préparations injectables dans leflacon.
•Retourner le flacon doucement jusqu'à complète dissolution. Ne passecouer.
La solution reconstituée doit être inspectée visuellement avant l'administration afin de détecter toute présence éventuelle de particules ou une décoloration. La solution reconstituée ne doit pas comporter de particules visibles et doit être transparente à légèrement opalescente. La couleur de la solution reconstituée doit être incolore à brun pâle. Ne pas utiliser si la solution reconstituée contient des particules visibles ou est trouble ou décolorée.
Instructions pour la dilution
Déterminer le volume de solution reconstituée requis sur la base d'une dose de 3,6 mg de trastuzumab emtansine/kg de poids corporel (voir rubrique Posologie et mode d'administration) :
Volume (mL) = Dose totale à administrer (poids corporel (kg) x dose (mg/kg))20 (concentration de la solution reconstituée en mg/mL)
La quantité appropriée de solution doit être prélevée du flacon et ajoutée dans une poche pour perfusion contenant 250 mL d'une solution de chlorure de sodium à 4,5 mg/mL (0,45 %) pour perfusion ou de chlorure de sodium à 9 mg/mL (0,9 %) pour perfusion. Une solution de glucose (5 %) ne doit pas être utilisée (voir rubrique Incompatibilités). Une solution de chlorure de sodium à 4,5 mg/mL (0,45 %) pour perfusion peut être utilisée sans un filtre en ligne en polyéthersulfone de 0,20 ou 0,22 microns. Si une solution de chlorure de sodium à 9 mg/mL (0,9 %) pour perfusion est utilisée pour la perfusion, un filtre en ligne en polyéthersulfone de 0,20 ou 0,22 microns est requis. Une fois la perfusion préparée, elle doit être administrée immédiatement. Ne pas congeler ou agiter la solution pour perfusion durant la conservation.
Élimination
Le produit reconstitué ne contient pas de conservateur et est à usage unique seulement. Eliminer toute quantité nonutilisée.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Médicament réservé à l’usage hospitalier.
Prescription réservée aux spécialistes en oncologie ou aux
médecins compétents en cancérologie.
Médicament nécessitant une surveillance particulière pendant
le traitement.
Poudre pour solution à diluer pour perfusion.
Poudre lyophilisée blanche à blanc cassé.
Kadcyla se présente dans un flacon de verre de type I de 20 mL (160 mg), muni d'un bouchon en élastomère butyle gris recouvert d'un film de fluororésine et scellé avec une capsule en aluminium avec un opercule en plastique violet.
Boîte de 1 flacon.