
WAYLIVRA 285 mg, solution injectable, boîte de 1 seringue préremplie de 1,50 ml
Retiré du marché le : 22/10/2019
Dernière révision : 21/06/2019
Taux de TVA : 0%
Laboratoire exploitant : AKCEA THERAPEUTICS FRANCE
Source :

WAYLIVRA est indiqué en complément d'un régime diététique chez les patients adultes atteints du Syndrome d'Hyperchylomicronémie Familial (SHCF) génétiquement confirmé et à risque élevé de pancréatite, chez qui la réponse au régime alimentaire et au traitement visant à réduire les triglycérides a été insuffisante.
Hypersensibilité à la/aux substance(s) active(s) ou à l'un des excipients mentionnés à la rubrique Composition.
Thrombocytopénie chronique ou persistante. Ne pas instaurer un traitement par WAYLIVRA chez des patients atteints de thrombocytopénie (numération plaquettaire < 140 000/mm3).
Thrombocytopénie
WAYLIVRA est associé à une réduction du nombre de plaquettes chez les patients atteints de SHCF, ce qui peut entraîner une thrombocytopénie (voir rubrique Effets indésirables). Des patients atteints de SHCF ont également été décrits comme présentant une thrombocytopénie intermittente. Les patients ayant un poidsplusfaible(inférieurà70kg)peuventêtredavantagesujetsàlathrombocytopéniependantqu'ils prennent WAYLIVRA. Un suivi attentif de la thrombocytopénie est important pendant le traitement par WAYLIVRA chez les patients atteints de SHCF (voir rubrique Posologie et mode d'administration). Les recommandations relatives aux ajustements de la fréquence de suivi et de l'administration de WAYLIVRA sont indiquées dans le tableau 1.
L’arrêt de médicament anti-plaquettaire/AINS/anticoagulants doit être considéré si le taux de plaquettes est < 75 x 109 /L. Ces médicaments doivent être arrêtés si le taux est < 50 x 109 /L WAYLIVRA (voir rubriqueInteractions avec d'autres médicaments et autres formes d'interactions).
Les patients doivent avoir pour consigne de prévenir immédiatement leur médecin s'ils présentent des signes de saignement, ce qui peut comprendre des pétéchies, des ecchymoses spontanées, des saignements sous-conjonctivaux ou d'autres saignements inhabituels (y compris des saignements de nez, des saignements des gencives, des selles sanglantes ou des saignements menstruels anormalement abondants), une raideur de la nuque, des céphalées intenses atypiques ou tout saignement prolongé.
Taux de LDL-C
Avec le traitement par WAYLIVRA, les taux de LDL-C peuvent augmenter, mais ils demeurent habituellement dans la plage normale.
Toxicité rénale
Une toxicité rénale a été observée après l'administration de WAYLIVRA et d'autres oligonucléotides antisens par voie sous-cutanée et intraveineuse. Il est recommandé d'effectuer une surveillance trimestrielle de la néphrotoxicité au moyen d'une bandelette d'analyse d'urine de routine. Dans le cas d'une évaluation positive, une évaluation plus large de la fonction rénale, y compris la créatinine et un prélèvement sur 24 heures pour quantifier la protéinurie et évaluer la clairance de la créatinine, devrait être effectuée. Le traitement doit être interrompu si : une protéinurie de ≥ 500 mg/24 heures est enregistrée, ou une augmentation de la créatinine sérique ≥ 0,3 mg/dL (26,5 µmol/L) qui est >LSN est enregistrée,ousilaclairancedelacréatinineestiméeparl'équationCKD-EPIest≤30mL/min/1,73m2. Il faut également interrompre le traitement pour tout symptôme clinique ou signe d'insuffisance rénale en attendant les évaluations de confirmationprécédentes.
Hépatotoxicité
Des élévations des enzymes hépatiques ont été observées après l'administration d'autres oligonucléotides antisens administrés par voie sous-cutanée et intraveineuse. La surveillance de l'hépatotoxicité par les enzymes hépatiques sériques et la bilirubine devrait être évaluée trimestriellement. Le traitement doit être interrompu s'il y a une seule augmentation du taux d'ALT ou d'AST > 8 x LSN, ou une augmentation > 5 x LSN, qui persiste pendant 2 semaines, ou une augmentation moindre du taux d'ALT ou d'AST associée à une bilirubine totale > 2 x LSN ou INR> 1.5. Letraitementdoitégalementêtreinterrompuencasdesymptômescliniquesoudesignesd'insuffisance hépatique oud'hépatite.
Immunogénicité et inflammation
Aucun signe de modification du profil d'innocuité ou de la réponse clinique n'a été associé à la présence d'anticorps anti-drogue. Si l'on soupçonne la formation d'anticorps anti-drogue ayant un effet cliniquement significatif, communiquer avec le titulaire de l'autorisation de mise en marché pour discuter de l'analyse des anticorps. La surveillance de l'inflammation devrait être évaluée au moyen d'une évaluation trimestrielle de la vitesse de sédimentation (VS).Teneur en sodium
Ce médicament contient moins de 1 mmol de sodium (23 mg) par dose de 300 mg ; autrement dit, il est dans l'absolu sans sodium.
Résumé du profil de sécurité
Dans les études cliniques menées chez des patients atteints de SHCF, les effets indésirables les plus fréquemment signalés au cours du traitement ont été une diminution de la numération plaquettaire (voir rubrique Mises en garde et précautions d'emploi), chez 40 % des patients au cours des études pivotales, et des réactions au point d'injection, chez 82 % des patients.
Tableau résumant les effetsindésirables
Letableau3présenteleseffetsindésirableslorsdel'étudepivotaucoursdelaquellelespatientsont reçu WAYLIVRA par voiesous-cutanée.
La fréquence des effets indésirables est définie selon la convention suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000) ; et fréquence inconnue (ne peut pas être estimée à partir des données disponibles). Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre décroissant de fréquence.
Tableau 3 : Résumé des effets indésirables dans l'étude APPROACH menée chez des patients atteints de SHCF (N=86)
| Classe de systèmes d'organes | Très fréquent (N,%) | Fréquent (N,%) |
| Affections hématologiques et du système lymphatique | Thrombocytopénie (10, 12 %) | Leucopénie (2, 2%) Éosinophilie (1 , 1 %) Purpura thrombopénique immunitaire (1, 1 %) Hématome spontané (1, 1 %) |
| Troubles du système Immunitaire | Immunisation (3, 3%) Hypersnsibilité (1, 1%)` Réaction de type maladie sérique (1, 1 %) | |
| Troubles du métabolisme et de la nutrition | Diabète sucré (1, 1 %) | |
| Troubles psychiatriques | Insomnie (1, 1%) | |
| Affections du système nerveux | Céphalées (8, 9%) Hypoesthésie (1, 1 %) Présyncope (1, 1%) Migraines ophtalmiques (1, 1%) Syncope (1, 1%) Tremblements (1, 1%) Vertiges (1, 1%) | |
| Affections oculaires | Hémorragie conjonctivale (1, 1%) Vision floue (1, 1%) | |
| Affections vasculaires | Hématome (1, 1%) Hypertension artérielle (1, 1%) Hémorragie (1, 1%) Bouffée de chaleur (1, 1%) | |
| Affections respiratoires, thoraciques et médiastinales | Épistaxis (3, 3%) Toux (1, 1 %) Dyspnée (2, 2 %) Congestion nasale (1, 1 %) Œdème pharyngé (1, 1 %) Respiration sifflante (1, 1 %) | |
| Affections gastro-intestinales | Nausées (8, 9%) Diarrhée (4, 5%) Bouche sèche (1, 1 %) Saignements gingivaux (1, 1 %) Hémorragie buccale (1, 1 %) Hypertrophie de la glande parotide (1, 1 %) Vomissements (4, 5 %) Douleurs abdominales (4, 5%) Distension abdominales (1, 1%) Dyspepsie (1, 1%) Gonflement des gencives (1, 1%) | |
| Affections de la peau et du tissu sous-cutané | Érythème (4,5 %) Prurit (4,5 %) Urticaire (3,3%) Hyperhidrose (2,2%) Éruptions cutanées (3,3 %) Pétéchies (1,1 %) Ecchymose (1, 1%) Sueurs nocturnes (1, 1%) Papule (1, 1%) Hypertrophie cutanée (1,1%) Gonflement du visage (1,1%) |
| Classe de systèmes d'organes | Très fréquent (N,%) | Fréquent (N,%) |
| Affections musculo- | Myalgie (8,9 %) | |
| squelettiques et systémiques | Arthralgie (6,7 %) | |
| Douleurs aux extrémités (5,6%) | ||
| Arthrite (2,2 %) | ||
| Douleurs dorsales (2,2 %) | ||
| Douleurs musculo-squelettiques | ||
| (2,2 %) | ||
| Douleurs aux cervicales (2,2%) | ||
| Spasmes musculaires (1,1%) | ||
| Raideur articulaire (1, 1%) | ||
| Myosite (1,1 %) | ||
| Douleur à la mâchoire (1,1 %) | ||
| Polymyalgie rhumatismale (1,1%) | ||
| Troubles | ||
| Affections du rein et des voies urinaires | Hématurie (1,1%) Protéinurie (1,1%) | |
| Troubles généraux et | Érythème au point d'injection | Asthénie (8,9 %) |
| anomalies au site | (67,78 %) | Fatigue (8,9 %) |
| d'administration | Douleur au point d'injection | Hématome au point d'injection |
| (38,44 %) | (7,8 %) | |
| Pâleur au point d'injection | Réaction au point d'injection (6,7 | |
| (37, 43 %) | %) | |
| Enflure au point d'injection | Urticaire au point d'injection (5,6 | |
| (25,29 %) | %) | |
| Prurit au point d'injection | Chaleur au point d'injection (5,6 | |
| (22,26 %) | %) | |
| Décoloration au point | Refroidissement (5, 6%) | |
| d'injection (19, 22%) | Pyrexie (4,5 %) | |
| Induration du site d'injection | Sécheresse au point d'injection | |
| (17, 20%) | (4,5 %) | |
| Ecchymoses au point | Hémorragie au point | |
| d'injection (10, 12 %) | d'injection (4,5 %) | |
| Œdème au point d'injection | Hypoesthésie au point d'injection | |
| (10, 12 %) | (4, 5%) | |
| Vésicules au point d'injection (3, 3 | ||
| %) | ||
| Malaise (2, 2%) | ||
| Sensation de chaleur (2, 2%) | ||
| Maladie pseudo-grippale (2, 2 %) | ||
| Inconfort au point d'injection (2, 2 | ||
| %) | ||
| Inflammation au point d'injection | ||
| (2, 2%) | ||
| Masse au point d'injection (2,2 %) | ||
| Douleur (2,2 %) | ||
| Paresthésie au point d'injection | ||
| (1, 1%) | ||
| Gale au point d'injection (1,1 %) | ||
| Papule au point d'injection (1, 1 | ||
| %) | ||
| Œdème (1, 1 %) | ||
| Douleurs thoraciques non | ||
| cardiaques (1, 1 %) | ||
| Hémorragie au site de ponction | ||
| veineux (1, 1 %) |
| Classe de systèmes d'organes | Très fréquent (N,%) | Fréquent (N,%) |
| Investigations | Diminution de la numération | Augmentation de la créatinine |
| plaquettaire (34 ,40 %) | sanguine (1, 1 %) | |
| Augmentation de l'urémie (1, | ||
| 1 %) | ||
| Diminution de la clairance rénale | ||
| de la créatinine (1, 1 %) | ||
| Augmentation du taux de | ||
| transaminases (1, 1 %) | ||
| Diminution du nombre de | ||
| globules blancs dans le sang (1, | ||
| 1 %) | ||
| Diminution de l'hémoglobinémie | ||
| (1, 1 %) | ||
| Augmentation des enzymes | ||
| hépatiques (1, 1 %) | ||
| Augmentation de l'INR (1, 1 %) | ||
| Lésions, intoxications et complications liées aux procédures | Contusion (3, 3 %) |
Description de certains effets indésirables
Thrombocytopénie
Dans l’étude pivot de phase 3 sur WAYLIVRA menée chez des patients atteints de SHCF (étude APPROACH), on a observé des réductions confirmées du nombre de plaquettes à des numérations en deçà de la normale (140 000/mm3 ) chez 75 % des patients atteints de SHCF traités par WAYLIVRA et 24 % des patients sous placebo ; des réductions confirmées à des numérations inférieures à 100 000/mm3 ont été observées chez 47 % des patients traités par WAYLIVRA contre aucun patient du bras placebo. Dans l’étude APPROACH et son extension en ouvert, les patients qui ont interrompu le traitement en raison de la numération plaquettaire comprenaient 3 patients dont la numération plaquettaire était inférieure à 25 x 109 /L, 2 dont la numération plaquettaire se situait entre 25 x 109 /L et 50 x 109 /L et 5 dont elle était comprise entre 50 x 109 /L et 75 x 109 /L. Aucun de ces patients n'a présenté d'hémorragie majeure et tous sont revenus à une numération plaquettaire normale après l'arrêt du traitement et l'administration de glucocorticostéroïdes lorsque cela était indiqué sur le plan médical.Immunogénicité
Lors des essais cliniques de phase 3 (COMPASS et APPROACH), 16 % et 30 % des patients traités par WAYLIVRA ont présenté un résultat positif à la recherche d’anticorps anti-médicament au cours des 6 mois et des 12 mois de traitement, respectivement. Aucun signe d’altération du profil de sécurité ou de la réponse clinique n’a été associé à la présence d’anticorps anti-médicament d’après les données limitées à long terme (voir rubriqueMises en garde et précautions d'emploi).
Réactions au site d'injection
Des réactions au site d’injection, définies comme étant toute réaction cutanée locale au site d’injection persistant plus de 2 jours, se sont produites après 82 % des patients traités par Waylivra 285 mg. Ces réactions locales étaient généralement légères et comprenaient habituellement au moins 1 des réactions suivantes : érythème, douleurs, prurit ou gonflement localisée. Les réactions au site d’injection ne se produisent pas avec toutes les injections et ont entraîné l’arrêt du traitement chez 1 patient de l’étude APPROACH.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté selon les modalités prévues dans le PUT.
AVANT l'instauration du traitement : mesure de la numération plaquettaire. Si la
numération plaquettaire est inférieure à 140 x 109
/L, une autre mesure doit être prise environ une
semaine plus tard pour réévaluer la situation. Si la numération plaquettaire reste inférieure à 140 x 109
/L
lors d'une seconde mesure, il ne faut pas initier le traitement.
SURVEILLANCE du traitement :
- Numération plaquettaire toutes les deux
semaines.
- Surveillance
trimestrielle de la néphrotoxicité au moyen d'une bandelette d'analyse d'urine de routine. Dans le cas
d'une évaluation positive, une évaluation plus large de la fonction rénale, y compris la créatinine et un
prélèvement sur 24 heures pour quantifier la protéinurie et évaluer la clairance de la créatinine, devrait
être effectuée.
- Surveillance trimestrielle de
l'hépatotoxicité par les enzymes hépatiques sériques et la bilirubine.
- Surveillance de l'inflammation au moyen d'une évaluation trimestrielle de la
vitesse de sédimentation (VS).
CONSULTER IMMEDIATEMENT LE MEDECIN en cas de :
- Saignements
inhabituels ou prolongés, comme de petites plaques rouges apparaissant sur la peau (également appelées
pétéchies), des ecchymoses inexpliquées, des saignements qui ne s'arrêtent pas ou des saignements de
nez, ou en cas de raideur dans la nuque ou de maux de tête inhabituels.
- Signes d'altération des reins tels que le gonflement des chevilles, jambes et pieds, une diminution
du volume des urines, un essoufflement, la sensation d'être malade, une sensation de confusion ou de
grande fatigue ou de somnolence.
EVITER la consommation d'alcool pendant le traitement.
En cas d'OUBLI d'une dose dans les 48 heures, administrer la dose oubliée
dès que possible. Passé 48 heures, administrer la dose oubliée à la
date de la prochaine injection prévue.
Grossesse
Il n'existe pas de données sur l'utilisation de WAYLIVRA chez la femme enceinte.
Les études effectuées chez l’animal n’ont pas mis en évidence d’effets délétères directs ou indirects sur la reproduction (voir rubrique Données de sécurité précliniques.).
Par mesure de précaution, il ne faut pas utiliser WAYLIVRA pendant la grossesse.
Allaitement
Dans les études non cliniques, les concentrations de WAYLIVRA dans le lait étaient très faibles chez des souris en lactation. Les données de pharmacodynamie et toxicologie disponibles ont montrées un taux d'excrétion très faible de volanesorsen dans le lait (voir section Données de sécurité précliniques). En raison de la faible biodisponibilité orale de volanesorsen, on considère qu'il est peu probable que ces faibles concentrations dans le lait entraînent une exposition systémique due à l'allaitement.
On ne sait pas si le WAYLIVRA ou ses métabolites sont excrétés dans le lait maternel.
Les données pharmacodynamiques/toxicologiques disponibles chez l'animal ont mis en évidence l'excrétion de WAYLIVRA dans le lait (voir rubrique Données de sécurité précliniques).
Un risque pour les nouveau-nés/nourrissons ne peut être exclu.
Par mesure de précaution, WAYLIVRA ne doit pas être utilisé chez les femmes qui allaitent.
La décision d'interrompre l'allaitement maternel ou d'interrompre / éviter de prendre le traitement doit tenir compte des avantages de l'allaitement pour l'enfant et du bénéfice du traitement pour la femme.
Fertilité
Aucune donnée clinique sur l'effet de WAYLIVRA sur la fertilité humaine n'est disponible. Le WAYLIVRA n'a eu aucun effet sur la fertilité chez la souris.
Aucune étude clinique d'interaction médicamenteuse n'a été réalisée.
On ne s'attend pas à des interactions pharmacocinétiques d'importance clinique entre WAYLIVRA et les substrats, inducteurs ou inhibiteurs des enzymes du cytochrome P450 (CYP) et les transporteurs de médicament. Dans les études cliniques, WAYLIVRA a été utilisé en association avec des fibrates et des huiles de poisson sans effet sur la pharmacodynamique ou la pharmacocinétique de Waylivra. Aucun effet indésirable lié à des interactions médicamenteuses n'a été signalé au cours du programme clinique avec Waylivra.
On ne connaît pas les effets de l'administration concomitante de WAYLIVRA avec de l'alcool ou des médicaments connus pour leur potentiel hépatotoxique (p. ex. le paracétamol). Si des signes et symptômes d'hépatotoxicité apparaissent, il faut cesser d'utiliser le médicament hépatotoxique.
Agents antithrombotiques et médicaments susceptibles d'abaisser la numération plaquettaire
On ignore si le risque de saignement est accru par l’utilisation concomitante de WAYLIVRA et d’agents antithrombotiques ou de médicaments pouvant réduire la numération plaquettaire ou nuire à la fonction plaquettaire. L'arrêt des antiplaquettaires, des AINS et des anticoagulants doit être envisagé pour les taux plaquettaires <75 x 109 /L et le traitement avec ces médicaments doit être arrêté aux taux plaquettaires <50 x 109 /L (voir rubriqueMises en garde et précautions d'emploi).
Posologie
Le traitement doit être instauré par un médecin expérimenté dans le traitement des patients atteints de SHCF et rester sous sa supervision. Avant d'instaurer un traitement par WAYLIVRA, les causes secondaires de l'hypertriglycéridémie (p. ex. diabète non maîtrisé, hypothyroïdie) doivent être exclues ou traitées de façon appropriée.
La dose initiale recommandée est de 285 mg dans 1,5 ml, injectée par voie sous-cutanée une fois par semaine pendant 3 mois. Après 3 mois, la fréquence des doses doit être réduite à 285 mg toutes les 2 semaines.
Le traitement doit être interrompu chez les patients qui présentent une réduction du taux de triglycérides <25% ou chez qu'il atteinte d'un taux de triglycérides sériques <22,6 mmol/L après 3 mois de traitement avec WAYLIVRA dosé à 285 mg par semaine aéchoué.
Après6moisdetraitementavecWAYLIVRA,uneaugmentationdelafréquenced'administrationà285 mg par semaine peut être considérée si la diminution du taux de triglycérides sériques n'est pas jugée comme suffisante par le prescripteur expérimenté en charge du patient et si le taux de plaquettes se trouve dans des valeurs normales. Si l'augmentation de dose à 285 mg par semaine n'apporte pas de réduction additionnelle significative du taux de triglycérides après 9 mois de traitement, la posologie devra de nouveau passer à 285 mg toutes les 2 semaines.
Les patients doivent être informés qu'ils doivent réaliser l'injection le même jour de la semaine, en suivant la fréquence d'administration établie par le médecin.
Siunedoseaétéomisedansles48heures,lepatientaurapourconsigned'administrerladoseoubliée dès que possible. Si on ne s'en aperçoit pas dans les 48 heures, il faut administrer la dose oubliée à la date de la prochaine injectionprévue.
Suivi des plaquettes et adaptation posologique
Avant l'instauration du traitement par Waylivra, la numération plaquettaire doit être mesurée. Si la numération plaquettaire est inférieure à 140 x 109/L, une autre mesure doit être prise environ une semaine plustard pourré évaluer la situation.Si la numération plaquettaire reste inférieure à 140x109/L lors d'une seconde mesure, il ne faut pas initier le Waylivra (voir sectionContre-indications).
Après le début du traitement, les patients doivent faire suivre leur taux de plaquettes toutes les deux semaines.
La posologie doit être corrigée en fonction des valeurs de laboratoire conformément au tableau suivant.
Pour toute interruption ou tout arrêt du traitement d’un patient en raison d'une thrombocytopénie grave, les bénéfices et les risques d'un retour au traitement après retour d’une numération plaquettaire ≥140 x 109 /L doivent être soigneusement étudiés. En cas d'arrêt du traitement, il faut consulter un hématologue avant de reprendre le traitement.
Tableau 1. Suivi et recommandations pour le traitement par Waylivra
| Numération plaquettaire (x109/L) | Dose (seringue préremplie 300 mg) | Fréquence de la surveillance plaquettaires |
| Normal (≥140) | Instauration du traitement : Toutes les semaines Après 3 mois : Toutes les 2 semaines | Toutes les 2 semaines |
| De 100 à 139 | Toutes les 2 semaines | Hebdomadaire |
| De 75 à 99 | Suspension du traitement pour au moins 4 semaines. Ré-initiation du traitement après obtention d'un taux de plaquettes ≥ 100x109/L. | Hebdomadaire |
| De 50 à <74a | Suspension du traitement pour au moins 4 semaines. Ré-initiation du traitement après obtention d'un taux de plaquettes ≥ 100x109/L. | Tous les 2 à 3 jours |
| <50 a, b | Arrêt du traitement, glucocorticoïdes recommandés | Quotidienne |
a : Voir la section Mises en garde et précautions d'emploi pour les recommandations concernant l'utilisation des antiplaquettaires, des AINS et des anticoagulants.
b:laconsultationd'unhématologueestnécessaireafind'évaluerleratiobénéfice/risquedelapoursuiteéventuelledutraitement avec WAYLIVRA.
Populations particulières
Personnes âgées
Aucun ajustement de la dose de départ n'est nécessaire chez les patients âgés. Les données cliniques concernant les patients âgés de 65 ans et plus sont limitées (voir rubriques Propriétés pharmacodynamiques et Propriétés pharmacocinétiques).
Patients atteints d'insuffisance rénale
WAYLIVRA n’a pas été étudié et doit être utilisé avec prudence chez les patients atteints d’insuffisance hépatique. Le médicament n'est pas métabolisé par le système enzymatique du cytochrome P450 dans le foie ; il est donc peu probable qu'un ajustement posologique soit nécessaire chez les patients atteints d'insuffisance hépatique.Population pédiatrique
La sécurité et l'efficacité de WAYLIVRA chez les enfants et les adolescents âgés de moins de 18 ans n'ont pas encore été établies. Aucune donnée n'est disponible.
Mode d'administration
WAYLIVRA est destiné à un usage sous-cutané. Ne pas administrer par voie intramusculaire ou intraveineuse.
Chaque seringue préremplie est à usage unique.
WAYLIVRAdoitêtreinspectévisuellementavantl'administration.Lasolutiondoitêtrelimpideetincolore ou jaunâtre. Si la solution est trouble ou contient des particules visibles, ne pas injecter le contenu et rapporter le produit à lapharmacie.
La première injection administrée par le patient ou le soignant doit être effectuée sous la supervision d'un professionnel de santé dûment qualifié. Les patients et/ou les soignants doivent être formés à l'administration de WAYLIVRA conformément à la notice d'information du patient.
La seringue préremplie de WAYLIVRA doit être amenée à température ambiante avant l'injection. Elle doit être sortie du réfrigérateur (2 à 8 °C) au moins 30 minutes avant utilisation. Il ne faut pas utiliser d'autres méthodes de réchauffement. Il est normal de voir une grande bulle d'air. Ne pas essayer de retirer la bulle d'air.Il est important d’effectuer une rotation des sites d’injection. Les sites d’injection comprennent l’abdomen, la partie supérieure de la cuisse ou l’extérieur du bras. Si l’injection est administrée dans le bras, elle doit être faite par une autre personne. Il faut éviter de réaliser l’injection au niveau de la taille et à d’autres endroits où des pressions ou des frottements dus aux vêtements peuvent se produire. Ne pas injecter WAYLIVRA dans les tatouages, les grains de beauté, les taches de naissance, les ecchymoses, les éruptions cutanées ou les zones où la peau est sensible, rouge, durcie, meurtrie, lésée, brûlée ou enflammée.
Pour consulter les directives relatives à la manipulation et l'élimination du produit, voir rubrique Instructions pour l'utilisation, la manipulation et l'élimination.
Durée de conservation :
30 mois
LeWAYLIVRApeutêtresortiduréfrigérateuretconservé,danssonemballaged'origine,àtempérature ambiante (inférieure à 30 °C) jusqu'à 6 semaines. Pendant cette période de 6 semaines, WAYLIVRA peut être conservé au réfrigérateur et à température ambiante (jusqu'à 30 °C), selon les besoins. Jeter WAYLIVRA immédiatement s'il n'a pas été utilisé dans les 6 semaines qui ont suivi sa sortie initiale du réfrigérateur.
Précautions particulières de conservation :À conserver au réfrigérateur, entre 2 °C et 8 °C, dans l'emballage d'origine à l'abri de la lumière. Ne pas congeler.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Il n'y a pas d'expérience clinique de surdosage de WAYLIVRA. En cas de surdosage, les patients doivent être suivis de près et recevoir un traitement symptomatique, le cas échéant.
Il est peu probable que l'hémodialyse soit bénéfique étant donné que WAYLIVRA est rapidement distribué dans les cellules.
Classe pharmacothérapeutique : {non encore attribué} Code ATC : {non encore attribué}
Mécanisme d'action
WAYLIVRA est un oligonucléotide antisens conçu pour inhiber la formation de l'apoC-III, une protéine reconnue pour réguler à la fois le métabolisme des triglycérides et la clairance hépatique des chylomicrons et d'autres lipoprotéines riches en triglycérides. La liaison sélective du WAYLIVRA à l'acide ribonucléique messager (ARNm) de l'apoC-III dans la région non traduite 3' à la position de paires de bases 489-508 entraîne la dégradation de l'ARNm. Cette liaison empêche la traduction de la protéine apoC-III, éliminant ainsi un inhibiteur de la clairance des triglycérides et facilitant le métabolisme par une voie indépendante de la lipoprotéine lipase (LPL).
Effets pharmacodynamiques
Effets de WAYLIVRA sur les paramètres lipidiques
Dans l’étude APPROACH, l’étude clinique de phase 3 sur WAYLIVRA menée chez des patients atteints de SHCF, WAYLIVRA a réduit les taux de triglycérides à jeun, de cholestérol total, de cholestérol nonHDL, d’apoC-III, d’apoB-48 et de triglycérides sous forme de chylomicrons, et a augmenté les taux de LDL-C, d’HDL-C et d’apoB(voir tableau4).
Tableau 4 : Valeurs de référence moyennes et variation en pourcentage des paramètres lipidiques entre le point de départ et la mesure d'évaluation principale (mois 3)
| Paramètre lipidique (g/l pour apoC-III, apoB, apoB- 48 ; mmol/l pour le cholestérol, les triglycérides) | Placebo (N = 33) | WAYLIVRA 285 mg (N = 33) | ||
| Valeur de référence | Variation en % | Valeur de référence | Variation en % | |
| Triglycérides | 24,3 | +24 % | 25,6 | -72 % |
| Cholestérol total | 7,3 | +13 % | 7,6 | -39 % |
| LDL-C | 0,72 | +7 % | 0,73 | +139 % |
| HDL-C | 0,43 | +5 % | 0,44 | +45 % |
| Cholestérol non HDL | 6,9 | +14 % | 7,1 | -45 % |
| ApoC-III | 0,29 | +6 % | 0,31 | -84 % |
| ApoB | 0,69 | +2 % | 0,65 | +20 % |
| ApoB-48 | 0,09 | +16 % | 0,11 | -75 % |
| Triglycérides sous forme de chylomicrons | 20 | +38 % | 22 | -77 % |
Électrophysiologie cardiaque
À une concentration en médicament égale à 4,1 fois la concentration plasmatique maximale (Cmax) du médicament à la dose maximale recommandée (300 mg par injection sous-cutanée), WAYLIVRA n'a pas prolongé l'intervalle QT corrigé (QTc) pour la fréquence cardiaque.
Efficacité et sécurité cliniques
Étude APPROACH chez des patients atteints de SHCF
L'étude APPROACH est un essai clinique multicentrique, randomisé, en double aveugle, contrôlé par placebo, d'une durée de 52 semaines, mené auprès de patients atteints de SHCF. Il a porté sur 66 patients atteints de SHCF et a évalué WAYLIVRA à la dose de 300 mg administrée par injection sous-cutanée (33 patients traités par WAYLIVRA et 33 patients sousplacebo).
Les principaux critères d'inclusion étaient un diagnostic de SHCF (hyperlipoprotéinémie de type 1) en combinaison avec des antécédents de chylomicronémie mis en évidence par la documentation du sérum lactescent ou de la mesure de la TG à jeun ≥ 880 mg/dl.
Diagnostic de SHCF avec documentation requise d'au moins un des éléments suivants :
a)Homozygote confirmé, hétérozygote composé ou hétérozygote double pour lesmutations connues de la perte de fonction dans les gènes responsables de la maladie de type 1 (tels que LPL, APOC2, GPIHBP1, ouLMF1)
b)Activité plasmatique LPL post-héparine de ≤ 20% de lanormale.
Les patients prenant Glybera dans les deux années précédant le dépistage ont été exclus de l'étude.
Dix-neuf des 33 patients du groupe WAYLIVRA ont terminé les 12 mois de traitement à l'étude. Treize de ces patients ont effectué un ajustement posologique ou une suspension de traitement pendant l'étude.
La moyenne d’âge était de 46 ans (entre 20 et 75 ans ; 5 patients ≥ 65 ans) ; 45 % étaient des hommes ; 80 % étaient blancs, 17 % étaient asiatiques et 3 % avaient une autre origine ethnique. L’indice de masse corporelle moyen était de 25 kg/m2 . Des antécédents documentés de pancréatite aiguë ont été signalés chez 76 % des patients, et des antécédents de diabète chez 15 % d’entre eux ; 21 % des patients présentaient des antécédents attestés de lipémie rétinienne et 23 % des patients présentaient des antécédents de xanthome éruptif. L’âge médian au moment du diagnostic était de 27 ans, 23 % des patients ne présentant pas de mutation génétique connue du SHCF.Au début de l’étude, 55 % des patients suivaient des traitements hypolipidémiants (48 % par des fibrates, 29 % par des huiles de poisson, 20 % par des inhibiteurs de l’HMG-CoA réductase), 27 % étaient sous analgésiques, 20 % prenaient des inhibiteurs de l’agrégation plaquettaire et 14 % des compléments alimentaires. Les traitements hypolipidémiants de fond sont demeurés constants tout au long de l’étude. Il était interdit aux patients de recevoir une aphérèse plasmatique dans les 4 semaines précédant la sélection ou pendant l’étude ; 11 % des patients avaient déjà reçu une thérapie génique pour un déficit en lipoprotéine lipase (c.-à-d. l’alipogène tiparvovec), en moyenne 8 ans avant le début de l’étude. Après une période de pré-inclusion sous régime diététique de 6 semaines, le taux moyen de triglycérides à jeun au départ était de 2 209 mg/dl (25,0 mmol/l). L’observance du régime diététique et de la restriction de la consommation d’alcool était renforcée par des séances de conseils périodiques au cours de l’étude.
Une analyse a posteriori a montré que WAYLIVRA a permis une réduction statistiquement significative des taux de triglycérides par rapport au placebo à la mesure d’évaluation principale de l’efficacité, définie comme la variation en pourcentage par rapport au niveau de référence au mois 3 du taux de triglycérides à jeun, en plus d’une incidence plus faible de pancréatite au cours de la période de traitement de 52 semaines (tableau 5).
À la mesure d’évaluation principale de l’efficacité, la différence de traitement entre WAYLIVRA et le placebo quant à la variation moyenne en pourcentage du taux de triglycérides à jeun était de -94 % (IC à 95 % : -122 % à -67 % ; p < 0,0001, avec une diminution de -77 % (IC à 95 % : -97 à -56) chez les patients recevant WAYLIVRA et une augmentation de 18 % (IC à 95 % : -4 à 39) chez les patients recevant le placebo (tableau 5).
Tableau 5 : Variation moyenne par rapport à la valeur de référence des taux de triglycérides à jeun dans l'essai de phase 3 contrôlé par placebo mené chez des patients atteints de SHCF au mois 3 (étude APPROACH)
| Placebo (N = 33) | WAYLIVRA (N = 33) | Différence relative en matière de variation par rapport au placebo | |
| Variation en pourcentage de la moyenne des moindres carrés (IC à 95 %) | +18 % (-4 à 39) | -77 % (-97 à -56) | -94 %* (-122 à -67) |
| Variation en valeur absolue de la moyenne des moindres carrés (IC à 95 %) mg/dl ou mmol/l | +92 (-301 à +486) mg/dl +1 (-3 à +5) mmol/l | -1 712 (-2 094 à -1 330) mg/dl -19 (-24 à -15) mmol/l | -1 804 (-2 306 à -1 302) mg/dl -20 (-26 à -15) mmol/l |
*valeur de p < 0,0001 (mesure d'évaluation principale de l'efficacité)
Différence = moyenne des moindres carrés de [variation en % pour WAYLIVRA - variation en % pour le placebo] (modèle ANCOVA)
Le début de la réduction a été rapide, avec une divergence par rapport au placebo dès 4 semaines et une réponse maximale à 12 semaines, la réduction cliniquement et statistiquement significative des taux de triglycérides étant maintenue pendant 52 semaines (figure 1). La variation en pourcentage du taux moyen de triglycérides à jeun était nettement différente entre les groupes WAYLIVRA et placebo à 3, 6 et 12 mois ; le groupe WAYLIVRA comprenait des patients qui n'avaient pas terminé le traitement, mais qui sont revenus effectuer des évaluations au cours de l'étude de 52 semaines. Il n'y avait pas de différences significatives en ce qui concerne l'effet du traitement entre les facteurs de stratification de présence ou d'absence de fibrates ou d'acides gras oméga- 3 concomitants.
Figure 1 : Variation en pourcentage de la moyenne des moindres carrés des taux de triglycérides à jeun dans l’essai de phase 3 mené chez des patients atteints de SHCF (étude APPROACH)
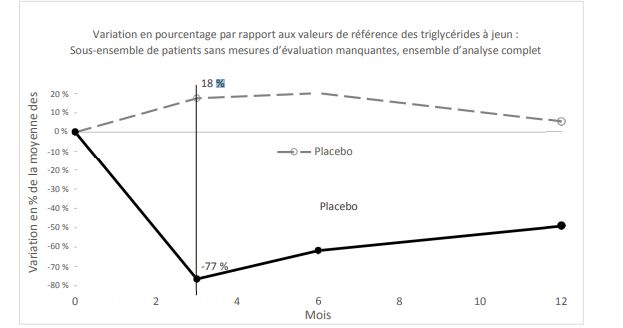
La courbe représente la variation en pourcentage de la moyenne des moindres carrés par rapport à la valeur de référence des taux de triglycérides à jeun en fonction des données observées.
Différence = moyenne des moindres carrés de [variation en % pour WAYLIVRA - variation en % pour le placebo] (modèle ANCOVA)
Valeur de p du modèle ANCOVA < 0,0001 au mois 3 (mesure d'évaluation principale de l'efficacité), au mois 6 et au mois 12.
Le tableau 6 présente d'autres résultats en termes d'efficacité pour les variations des taux de triglycérides. La plupart des patients recevant WAYLIVRA ont connu une réduction cliniquement significative des taux de triglycérides.
Tableau 6 : Résultats additionnels pour les variations des taux de triglycérides dans l'étude APPROACH (mesure d'évaluation principale au mois 3)
| Paramètre au mois 3a | Placebo (N = 31) | WAYLIVRA (N = 30) |
| Pourcentage de patientsb ayant des taux de triglycérides plasmatiques à jeun < 750 mg/dl (8,5 mmol/l)* | 10 % | 77 % |
| Pourcentage de patientsc affichant une réduction ≥ 40 % des taux de triglycérides à jeun** | 9 % | 88 % |
a La mesure d'évaluation au mois 3 était définie comme la moyenne des évaluations à jeun de la semaine 12 (jour 78) et de la semaine 13 (jour 85). S'il manquait 1 visite, c'est l'autre visite qui était utilisée pour la mesure d'évaluation.
b LedénominateurpourlecalculdupourcentageétaitlenombretotaldepatientsenFAS(FullAnalysisSet) ayant un taux de triglycérides à jeun de référence ≥ 750 mg/dl (ou 8,5 mmol/l) dans chaque groupe de traitement.
c Le dénominateur pour le calcul du pourcentage était le nombre total de patients dans chaque groupe de traitement.
* Valeur de p =0,0001
** Valeur de p <0,0001
Valeurs de p du modèle de régression logistique avec traitement, présence de pancréatite et présence simultanée d'acides gras oméga-3 et/ou de fibrates comme facteurs, et taux de triglycérides à jeun de référence après transformation logarithmique comme covariables.
Dans l'étude APPROACH, l'incidence numérique de la pancréatite chez les patients traités par WAYLIVRA était plus faible comparativement au groupe placebo (4 événements chez 3 patients du groupe placebo composé de 33 patients contre 1 événement chez 1 patient, du groupe WAYLIVRA composé de 33 patients).
Une analyse des patients ayant des antécédents de pancréatite récidivante (≥ 2 événements au cours des 5 années précédant le jour 1 de l’étude) a révélé une réduction significative des crises de pancréatite chez les patients traités par le WAYLIVRA comparativement aux patients sous placebo (p = 0,0242). Dans le groupe WAYLIVRA, sur les 7 patients qui avaient eu 24 crises de pancréatite établies au cours des 5 années antérieures, aucun des patients n’a connu d’attaque de pancréatite au cours de la période de traitement de 52 semaines. Dans le groupe placebo, sur les 4 patients qui avaient eu 17 crises de pancréatite établies au cours des 5 années antérieures, 3 patients ont connu 4 crises de pancréatite au cours de la période de traitement de 52 semaines.
Étude long terme ouverte chez des patients atteints de SHCF
Les données les plus récentes de l'étude CS7 en cours sont présentées au tableau 6. La variation en pourcentage de la TG à jeun entre la période de référence de l'étude indicielle et le troisième mois de l'étude ouverte pour les patients traités par APPROACH- et CS16-WAYLIVRA était de -49,2 % et -64,9 %, respectivement. Le changement en pourcentage de la TG à jeun selon l'indice de l'étude d’extension APPROACH WAYLIVRA était de -54,8% et de -35,1 %, respectivement, au 6e et au 12e mois, chez les patients traités par Waylivra 285 mg.| Time Point | Groupe naïf à l'égard du traitement (Étude ouverte, N=51) | APPROACH-WAYLIVRA (N=14) | CS16-WAYLIVRA (N=3) | ||||||
| n | Valeur observée | % de variation par rapport à la valeur d'inclusion dans l'étude CS7 | n | Valeur observée | % de variationpar rapport à la valeur d'inclusion dansl'étude APPROACH | n | Valeur observée | % change from Baseline in CS16 | |
| Valeur à | 2341 | 2641 | 2288 | ||||||
| l'inclusiona | 51 | (1193, | - | 14 | (1228, 328) | - | 3 | (1524, | - |
| 167) | 880) | ||||||||
| Mois 3 | 47 | 792 (589, 91) | -60.4 (36.459.8 (37.0, 5.64) | 14 | 1266 (812, 217) | -49.2 (34.8, 9.3) | 3 | 855 (651, 376) | -64.9 (9.1, 5.3) |
| Mois 6 | 979 | 1248 | -54.8 | 1558 | |||||
| (683, 107) | -46.45.5 | (927, 257) | (23.8, 6.6) | (193, | -39.1 (26.2, | ||||
| 49 | (42.9, 6 | 13 | 23 | 137)1215 | 18.543.0 | ||||
| (34.0, 5.3.1) | (610, | (19.7, 11.4) | |||||||
| 352) | |||||||||
| Mois 12 | 39 | 1287 (904, 213) | -34.9(40.8, 931.6(44.6, 7.1) | 1012 | 1411 (1143, 361)1670 (1198, 346) | -4735.1 (45.6, 13.2 (37.1, 11.7) | 03 | NC1369 (897, 518) | NC-39.9 (34.2, 19.7) |
| Mois 15 | 1071 | -30.1 (45.9, | 1751 (1850, | -6126.5 (57.4 | |||||
| 22 | (560, 211) | 1736.4 (41.0, | 310 | 1068)1886 (1219, | (29.5, 17.0, | 0 | NC | NC | |
| 8.7) | 386) | 18.1) | |||||||
| Mois 18 | 9 | 884 (826, 477) | -31.3 (8.4, 4.938.7 (42.1, 14.0) | 17 | 2391713 (1122, 424) | -82.0-38.4 (32.2, 12.2) | 0 | NC | NC |
a Les valeurs de référence pour le groupe n'ayant jamais été traité ont été tirées de l'étude ouverte CS7 et les valeurs de référence pour les groupes APPROACH-WAYLIVRA et CS16-WAYLIVRA ont été tirées de l'étude index respective.
NC = non calculé
Personnes âgées
Les études cliniques comprenaient 4 patients atteints de SHCF âgés de 65 ans traités par WAYLIVRA
dans le cadre d’études de contrôle randomisées (EC2, 1 patient ; EC6, 3 patients) et 3 patients âgés de
65 ans et plus dans l’étude en ouvert (EC7). Aucune différence globale en termes de sécurité ou
d’efficacité n’a été observée entre ces patients et des patients plus jeunes.
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec WAYLIVRA dans un ou plusieurs sous-groupes de la population pédiatrique pour le traitement du Syndrome d'Hyperchylomicronémie Familiale (voir rubrique Posologie et mode d'administrationpour les informations concernant l'usage pédiatrique).
Absorption
Après injection sous-cutanée, les concentrations plasmatiques maximales de WAYLIVRA sont généralement atteintes en 2 à 4 heures. La biodisponibilité absolue du WAYLIVRA après une administration sous-cutanée unique est d'environ 80 % (probablement plus élevée car une ASC de 0 à 24 heures a été utilisée et que la demi-vie du WAYLIVRA est supérieure à 2 semaines).
Après l’administration d’une dose de 300 mg une fois par semaine à des patients atteints de SHCF, la moyenne géométrique estimée de la Cmax à l’état d’équilibre est de 8,92 µg/ml, l’ASC0-168h est de 136 µg*h/ml et le Cmin est de 127 ng/ml. Un autre schéma posologique de 300 mg de WAYLIVRA toutes les deux semaines donne une Cmin,éé d’environ 58,0 ng/ml, avec une Cmax et une ASC semblables par rapport au schéma posologique à raison d’une fois par semaine
Distribution
Le WAYLIVRA était rapidement et largement distribué aux tissus après administration SC ou IV chez toutes les espèces évaluées. Le volume de distribution à l’état d’équilibre (Véé) estimé chez des patients atteints de SHCF est de 330 l. Le WAYLIVRA est fortement lié aux protéines plasmatiques humaines (> 98 %) et la liaison est indépendante de la concentration.
Des études in vitro montrent que WAYLIVRA n’est pas un substrat ou un inhibiteur de la glycoprotéine P (P-gp), de la protéine de résistance au cancer du sein (BCRP), des polypeptides de transport d’anions organiques (OATP1B1, OATP1B3), de la pompe d’exportation des sels biliaires (BSEP), des transporteurs de cations organiques (OCT1, OCT2) ou des transporteurs d’anions organiques (OAT1, OAT3).Biotransformation
Le WAYLIVRA n'est pas un substrat pour le métabolisme dépendant des CYP et est métabolisé dans les tissus par les endonucléases pour former des oligonucléotides plus courts qui sont ensuite des substrats d'un autre processus métabolique par les exonucléases.
Le WAYLIVRA sous sa forme inchangée est l'élément circulant prédominant.
Des études in vitro indiquent que WAYLIVRA n'est pas un inhibiteur du CYP1A2, du CYP2B6, du CYP2C8, du CYP2C9, du CYP2C19, du CYP2D6, du CYP2E1 ou du CYP3A4, ni un inducteur du CYP1A2, du CYP2B6 ou du CYP3A4.
Élimination
L'élimination implique à la fois le métabolisme dans les tissus et l'excrétion dans l'urine. La quantité du médicament d'origine retrouvée dans les urines était limitée chez l'homme, moins de 3 % de la dose sous-cutanée administrée étant récupérée dans les 24 heures suivant l'administration. Le médicament d'origine et les métabolites ayant une chaîne raccourcie de cinq à sept nucléotides (5 à 7-mer) représentaientenviron26%et55%desoligonucléotidesretrouvésdansl'urine,respectivement.Après administration sous-cutanée, la demi-vie d'élimination terminale est d'environ 2 à 5semaines.
Chez l’animal, l’élimination du WAYLIVRA a été lente et s’est produite principalement par excrétion urinaire, ce qui reflète une clairance plasmatique rapide principalement dans les tissus. Le WAYLIVRA et des métabolites oligonucléotidiques plus courts (principalement des métabolites de 7-mer (issus soit de délétions en 3’, soit de délétions en 5’)) ont été identifiés dans l’urine humaine.
Linéarité/non-linéarité
La pharmacocinétique du WAYLIVRA unidose et à doses multiples chez des volontaires sains et des patients atteints d'hypertriglycéridémie a montré que la Cmax du WAYLIVRA est proportionnelle à la dose sur une plage posologique de 100 à 400 mg et que l'ASC est légèrement plus que proportionnelle à la dose sur la même plage posologique.
L'état d'équilibre a été atteint environ 3 mois après l'instauration du traitement par WAYLIVRA. On a observé une accumulation de la Cmin (de 7 à 14 fois) et peu ou pas d'augmentation de la Cmax ou de l'ASC après une administration hebdomadaire SC à une dose de 200 à 400 mg. Une certaine accumulation de l'ASC et de la Cmax a été observée pour la dose de 50 à 100 mg. Étant donné que la dose administrée sera de 300 mg toutes les deux semaines, ou 142,5 mg par semaine, on s'attend à une faible augmentation de la Cmax ou de l'ASC si plusieurs doses sont administrées en contexte clinique.
Populations particulières
Insuffisance rénale
Une étude pharmacocinétique réalisée dans la population laisse penser qu'une insuffisance rénale légère ou modérée n'a aucun effet cliniquement pertinent sur l'exposition systémique au WAYLIVRA. Aucune donnée n'est disponible chez des patients atteints d'insuffisance rénale grave.
Insuffisance hépatique
La pharmacocinétique du WAYLIVRA chez des patients atteints d'insuffisance hépatique est inconnue.
Âge, sexe, poids et origine ethnique
Selonl'étudepharmacocinétiqueréaliséedanslapopulation,l'âge,lepoids,lesexeoul'origineethnique n'ont aucun effet cliniquement pertinent sur l'exposition auWAYLIVRA.
Formation d'anticorps anti-WAYLIVRA modifiant la pharmacocinétique
La formation d'anticorps qui se lient au WAYLIVRA semble avoir multiplié par 2 à 19 la Cmin totale.
WAYLIVRA n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, de génotoxicité et de toxicité des fonctions de reproduction et de développement, n’ont pas révélé de risque particulier pour l’homme.
Des réductions du nombre de plaquettes en fonction de la dose et du temps ont été observées dans les études à doses répétées chez le singe Cynomolgus. La diminution a été graduelle et autonome, et n’a pas atteint des taux défavorables. Chez le singe, une thrombocytopénie grave a été observée chez des groupes traités par le médicament à une dose ≥ 6 mg/kg/semaine (environ l’équivalent de l’exposition chez l’homme) au cours d’une étude de 9 mois et a également été observée dans les études cliniques. La diminution du nombre de plaquettes n’était pas marquée et la numération plaquettaire a baissé à moins de 50 000 cellules/µl. La numération plaquettaire s’est rétablie après l’arrêt du traitement, mais a de nouveau diminué en deçà de 50 000 cellules/µl après la reprise du traitement chez certains singes. On a également observé une diminution du nombre de plaquettes dans des études à doses répétées chez les rongeurs. Les autres effets indésirables observés dans les études de toxicité à doses répétées ne sont pas considérés comme étant pertinents pour l’homme. On ne connaît pas encore le mode d’action pour la thrombocytopénie observée.
Dans les études non cliniques, les concentrations de WAYLIVRA dans le lait étaient très faibles chez des souris en lactation. Les concentrations dans le lait maternel des souris étaient plus de 800 fois plus faibles que les concentrations effectives dans les tissus du foie maternel. En raison de la faible biodisponibilité orale du Waylivra 285 mg, on considère qu’il est peu probable que ces faibles concentrations dans le lait entraînent une exposition systémique due à l’allaitement (voir rubrique Fertilité, grossesse et allaitement).
WAYLIVRAdoitêtreinspectévisuellementavantl'administration.Lasolutiondoitêtrelimpideetincolore ou jaunâtre. Si la solution est trouble ou contient des particules visibles, ne pas injecter le contenu et rapporter le produit à lapharmacie.
N'utiliser chaque seringue préremplie qu'une seule fois, puis la placer dans un récipient pour objets pointus ou tranchants en vue de son élimination conformément aux directives communautaires.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Médicament
soumis à prescription hospitalière réservée aux spécialistes en
endocrinologie diabètologie-nutrition, hépato-gastro-entérologie, ou
cardiologie.
Médicament nécessitant une surveillance particulière pendant le traitement
Solution injectable (injection).
Solution limpide, incolore ou jaunâtre.
WAYLIVRA (volanesorsen) est fourni dans une seringue unidose préremplie en verre de type I, de 2,25 ml, munie d'un bouchon en caoutchouc chlorobutyle siliconé et d'une aiguille fixe avec protection, remplie pour administrer 1,5 ml de solution.
Présentation individuelle d'une seringue préremplie (1 boite de 1 seringue)