
ALUNBRIG 90 mg, comprimé pelliculé, boîte de 7
Retiré du marché le : 03/07/2020
Dernière révision : 15/09/2021
Taux de TVA : 2.1%
Laboratoire exploitant : TAKEDA FRANCE
Source :

Alunbrig est
indiqué en monothérapie pour le traitement des patients adultes atteints d'un
cancer bronchique non à petites cellules (CBNPC) avancé présentant un
réarrangement du gène ALK (ALK- positif) non précédemment traités par un
inhibiteur de tyrosine kinase ciblant la mutation ALK+.
Alunbrig est
indiqué en monothérapie pour le traitement des patients adultes atteints de
CBNPC avancé ALK-positif et prétraités par crizotinib.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Effets indésirables pulmonaires
Des effets indésirables pulmonaires sévères, menaçant le pronostic vital et fatals, y compris ceux accompagnés de signes évoquant une PID/pneumopathie inflammatoire, peuvent survenir chez les patients traités par Alunbrig (voir rubrique Effets indésirables).
La plupart des effets indésirables pulmonaires ont été observés au cours des 7 premiers jours de traitement. Les effets indésirables pulmonaires de grade 1-2 ont disparu à l'arrêt du traitement ou après une modification de la posologie. Un âge avancé et un délai court (moins de 7 jours) entre la dernière prise de crizotinib et la première prise d'Alunbrig étaient indépendamment associés à une augmentation du taux de ces effets indésirables pulmonaires. Ces facteurs doivent être pris en considération lors de l'instauration d'un traitement par Alunbrig. Les patients ayant des antécédents de PID ou de pneumopathie inflammatoire induite par les médicaments étaient exclus de l'étude pivotale.
Certains patients ont développé une pneumopathie inflammatoire ultérieurement au cours du traitement par Alunbrig.
Les patients doivent faire l'objet d'une surveillance afin de détecter l'apparition ou l'aggravation de symptômes respiratoires (par ex. dyspnée, toux, etc.), en particulier au cours de la première semaine de traitement. Si un patient présente des signes de pneumopathie inflammatoire accompagnés d'une aggravation des symptômes respiratoires, il doit être rapidement examiné. Si une pneumopathie inflammatoire est suspectée, le traitement par Alunbrig doit être suspendu et le patient examiné à la recherche d'autres causes pour ces symptômes (par ex. embolie pulmonaire, progression tumorale et pneumonie infectieuse). La posologie doit être modifiée en conséquence (voir rubrique Posologie et mode d'administration).
Hypertension
Une hypertension est survenue chez des patients traités par Alunbrig (voir rubrique Effets indésirables).
La pression artérielle doit être mesurée régulièrement pendant le traitement par Alunbrig. L'hypertension doit être traitée conformément aux recommandations en vigueur de contrôle de la pression artérielle. La fréquence cardiaque doit être surveillée plus fréquemment en cas de prise concomitante d'un médicament connu pour provoquer une bradycardie si ce dernier ne peut être évité. En cas d'hypertension sévère (grade ≥ 3), le traitement par Alunbrig doit être suspendu jusqu'à résolution à une hypertension de grade 1 ou aux valeurs tensionnelles initiales. La posologie doit être adaptée en conséquence (voir rubrique Posologie et mode d'administration).
Bradycardie
Une bradycardie est survenue chez des patients traités par Alunbrig (voir rubrique Effets indésirables). La prudence s'impose lors de l'administration d'Alunbrig en association avec des agents bradycardisants. La fréquence cardiaque et la pression artérielle doivent être surveillées régulièrement.
En cas de bradycardie symptomatique, le traitement par Alunbrig doit être suspendu et les médicaments concomitants connus pour provoquer une bradycardie doivent être évalués.Dès la résolution, la posologie doit être modifiée en conséquence (voir rubrique Posologie et mode d'administration). En cas de bradycardie menaçant le pronostic vital, si aucun médicament concomitant contribuant à la bradycardie n'a été identifié ou en cas de récidive, le traitement par Alunbrig doit être interrompu (voir rubrique Posologie et mode d'administration).
Troubles visuels
Des effets indésirables à type de troubles visuels sont survenus chez des patients traités par Alunbrig (voir rubrique Effets indésirables). Il doit être conseillé aux patients de signaler tout symptôme visuel. En cas d'apparition ou d'aggravation de symptômes visuels sévères, un examen ophtalmologique et une diminution de la posologie doivent être envisagés (voir rubrique Posologie et mode d'administration).
Augmentation des taux de créatine phosphokinase (CPK)
Des augmentations du taux de CPK sont apparues chez des patients traités par
Alunbrig (voir rubrique Effets indésirables). Il doit être conseillé aux
patients de signaler toute douleur, sensibilité douloureuse ou faiblesse
musculaire inexpliquée. Les taux de CPK doivent être mesurés régulièrement
pendant le traitement par Alunbrig. Selon la sévérité de l'augmentation des taux
de CPK, et si elle est associée d'une faiblesse ou des douleurs musculaires, le
traitement par Alunbrig doit être suspendu et la posologie modifiée en
conséquence (voir rubrique Posologie et mode d'administration).
Augmentations des enzymes pancréatiques
Des augmentations de l'amylase et de la lipase sont apparues chez des patients traités par Alunbrig (voir rubrique Effets indésirables). La lipase et l'amylase doivent être mesurées régulièrement pendant le traitement par Alunbrig. Selon la sévérité des anomalies biologiques, le traitement par Alunbrig doit être suspendu et la dose modifiée en conséquence (voir rubrique Posologie et mode d'administration).
Hépatotoxicité
Des augmentations des enzymes hépatiques (aspartate aminotransférase, alanine aminotransférase) et de bilirubine sont survenus chez des patients traités par Alunbrig (voir rubrique Effets indésirables). Un bilan de la fonction hépatique, notamment l'ASAT, l'ALAT et la bilirubine totale, doit être réalisé avant
l'initiation du traitement par Alunbrig, puis toutes les 2 semaines pendant les 3 premiers mois de traitement. Ensuite, une surveillance régulière est recommandée. Selon la sévérité des anomalies biologiques, le traitement doit être suspendu et la posologie modifiée en conséquence (voir rubrique Posologie et mode d'administration).
Hyperglycémie
Des augmentations de la glycémie sont apparues chez des patients traités par Alunbrig. La glycémie à jeun doit être mesurée avant l'initiation du traitement par Alunbrig et régulièrement surveillée par la suite. Un traitement hypoglycémiant doit être instauré ou optimisé si besoin. En l'absence d'un contrôle glycémique adéquat avec une prise en charge médicale adaptée, le traitement par Alunbrig doit être suspendu jusqu'à obtention d'un contrôle glycémique adéquat ; dès l'obtention d'un contrôle glycémique satisfaisant, le traitement peut être repris au palier de dose inférieur comme indiqué dans le tableau 1 ou le traitement par Alunbrig peut être arrêté définitivement.
Interactions médicamenteuses
L'utilisation concomitante d'Alunbrig et d'inhibiteurs puissants du CYP3A doit être évitée. Si l'utilisation concomitante d'inhibiteurs puissants du CYP3A ne peut être évitée, la posologie d'Alunbrig doit être réduite de 180 mg à 90 mg ou de 90 mg à 60 mg. Après l'arrêt d'un inhibiteur puissant du CYP3A, le traitement par Alunbrig doit être repris à la posologie tolérée avant l'initiation du traitement par inhibiteur puissant du CYP3A.
L'utilisation concomitante d'Alunbrig et d'inducteurs puissants et modérés du CYP3A doit être évitée (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Fertilité
Il doit être conseillé aux femmes en âge de procréer d'utiliser une méthode efficace de contraception non hormonale pendant toute la durée du traitement par Alunbrig et pendant au moins 4 mois après l'arrêt du traitement. Il doit être conseillé aux hommes dont les partenaires féminines sont en âge de procréer d'utiliser une méthode efficace de contraception pendant toute la durée du traitement par Alunbrig et pendant au moins 3 mois après l'arrêt du traitement (voir rubrique Grossesse et allaitement).
Lactose
Alunbrig contient du lactose monohydraté. Les patients présentant une intolérance au galactose, un déficit total en lactase ou un syndrome de malabsorption du glucose et du galactose (maladies héréditaires rares) ne doivent pas prendre ce médicament.
Résumé du profil de sécurité
Les effets indésirables les plus fréquents (≥ 25 %) rapportés chez les patients traités par Alunbrig à la dose recommandée étaient : augmentation de l'ASAT, augmentation du taux de CPK, hyperglycémie, augmentation du taux de lipase, hyperinsulinémie, diarrhée, augmentation de l'ALAT, augmentation du taux d'amylase, anémie, nausée, fatigue, hypophosphatémie, diminution des lymphocytes, toux, augmentation du taux de phosphatase alcaline, éruption cutanée, augmentation du temps de céphaline activé (TCA), myalgie, céphalée, hypertension, diminution des globules blancs, dyspnée et vomissements.
Les effets indésirables graves les plus fréquents (≥ 2 %) rapportés chez les patients traités par Alunbrig à la dose recommandée, outre les événements associés à la progression de la tumeur, étaient : pneumonie, pneumopathie inflammatoire, dyspnée et fièvre.
Liste des effets indésirables
Les données décrites ci-dessous reflètent l’exposition à Alunbrig au schéma posologique recommandé dans trois essais cliniques : un essai de phase III (ALTA 1L) mené chez des patients atteints d'un CBNPC ALK-positif avancé non précédemment traités par un inhibiteur de tyrosine kinase ciblant la mutation ALK+(N = 136), un essai de phase II (ALTA) mené chez des patients atteints d’un CBNPC ALK-positif traités par Alunbrig et ayant précédemment progressé sous crizotinib (N = 110) et un essai d’escalade de dose de phase I/II mené chez des patients atteints de tumeurs malignes avancées (N = 28). Dans ces études, la durée médiane d’exposition chez les patients recevant Alunbrig au schéma posologique recommandé était de 21,8 mois.
Les effets indésirables rapportés sont présentés dans le tableau 3 par classe de système d’organe, terme préférentiel et fréquence. Les catégories de fréquence sont : très fréquent (≥ 1/10), fréquent (≥ 1/100 à < 1/10) et peu fréquent (≥ 1/1 000 à < 1/100). Dans chaque groupe de fréquence, les effets indésirables sont présentés par ordre de fréquence.
Tableau 3 : Effets indésirables rapportés chez des patients traités par Alunbrig (selon les critères CTCAE (Common Terminology Criteria for Adverse Events) version 4.03) au schéma posologique de 180 mg (N = 274)
|
Classe de système d’organe |
Catégorie de fréquence |
Effets indésirables† tous grades |
Effets indésirables de Grade 3-4 |
|
Infections et infestations |
Très fréquent |
Pneumoniea,b Infection des voies respiratoires supérieures |
|
|
Fréquent |
|
Pneumoniea |
|
|
Affections hématologiques et du système lymphatique |
Très fréquent |
Anémie Diminution des lymphocytes Augmentation du TCA Diminution des globules blancs Diminution des neutrophiles |
Diminution des lymphocytes |
|
Fréquent |
Diminution du taux de plaquettes |
Augmentation du TCA Anémie |
|
|
Peu fréquent |
|
Diminution du nombre de neutrophiles |
|
|
Troubles du métabolisme et de la nutrition |
Très fréquent |
Hyperglycémie Hyperinsulinémiec Hypophosphatémie Hypomagnésémie Hypercalcémie Hyponatrémie Hypokaliémie Diminution de l’appétit |
|
|
Fréquent |
|
Hypophosphatémie Hyperglycémie Hyponatrémie Hypokaliémie Diminution de l’appétit |
|
|
Affections psychiatriques |
Fréquent |
Insomnie |
|
|
Affections du système nerveux |
Très fréquent |
Céphaléed Neuropathie périphériquee Sensations vertigineuses |
|
|
Fréquent |
Atteinte de la mémoire Dysgueusie |
Céphaléed Neuropathie périphériquee |
|
|
Peu fréquent |
|
Sensations vertigineuses |
|
|
Affections oculaires |
Très fréquent |
Troubles visuelsf |
|
|
Fréquent |
|
Troubles visuelsf |
|
|
Affections cardiaques |
Fréquent |
Bradycardieg Allongement de l’intervalle QT à l’ECG Tachycardieh Palpitations |
Allongement de l’intervalle QT à l’ECG |
|
Peu fréquent |
|
Bradycardieg |
|
|
Affections vasculaires |
Très fréquent |
Hypertensioni |
Hypertensioni |
|
Affections respiratoires, |
Très fréquent |
Toux Dyspnéej |
|
|
Classe de système d’organe |
Catégorie de fréquence |
Effets indésirables† tous grades |
Effets indésirables de Grade 3-4 |
|
thoraciques et médiastinales |
Fréquent |
Pneumopathie inflammatoirek |
Pneumopathie inflammatoirek Dyspnéej |
|
Affections gastro- intestinales |
Très fréquent |
Augmentation du taux de lipase Diarrhée Augmentation du taux d’amylase Nausée Vomissements Douleurs abdominalesl Constipation Stomatitem |
Augmentation du taux de lipase |
|
Fréquent |
Sécheresse buccale Dyspepsie Flatulences |
Augmentation du taux d’amylase Nausée Douleurs abdominalesl Diarrhée |
|
|
Peu fréquent |
Pancréatite |
Vomissements Stomatitem Dyspepsie Pancréatite |
|
|
Affections hépatobiliaires |
Très fréquent |
Augmentation de l’ASAT Augmentation de l’ALAT Augmentation du taux de phosphatase alcaline |
|
|
Fréquent |
Augmentation du taux de la lactate déshydrogénase sanguine Hyperbilirubinémie |
Augmentation de l’ALAT Augmentation de l’ASAT Augmentation du taux de phosphatase alcaline |
|
|
Peu fréquent |
|
Hyperbilirubinémie |
|
|
Affections de la peau et du tissu sous-cutané |
Très fréquent |
Eruption cutanéen Prurito |
|
|
Fréquent |
Sécheresse cutanée Réaction de photosensibilité |
Eruption cutanéen Réaction de photosensibilité |
|
|
Peu fréquent |
|
Sécheresse cutanée Prurito |
|
|
Affections musculo- squelettiques et systémiques |
Très fréquent |
Augmentation du taux de la CPK sanguine Myalgiep Arthralgie |
Augmentation du taux de la CPK sanguine |
|
Fréquent |
Douleurs thoraciques musculo- squelettiques Douleurs aux extrémités Rigidité musculo-squelettique |
|
|
|
Peu fréquent |
|
Douleurs aux extrémités Douleurs thoraciques musculo- squelettiques Myalgiep |
|
|
Affections du rein et des voies urinaires |
Très fréquent |
Augmentation de la créatinine sanguine |
|
|
Troubles généraux et anomalies au |
Très fréquent |
Fatigueq Œdèmer Fièvre |
|
|
Classe de système d’organe |
Catégorie de fréquence |
Effets indésirables† tous grades |
Effets indésirables de Grade 3-4 |
|
site d'administration |
Fréquent |
Douleurs thoraciques non cardiaques Gêne thoracique Douleurs |
Fatigueq |
|
Peu fréquent |
|
Fièvre Œdèmer Douleurs thoraciques non cardiaques |
|
|
Investigations |
Fréquent |
Augmentation du cholestérol sanguins Perte de poids |
|
|
Peu fréquent |
|
Perte de poids |
|
|
† Les fréquences des effets indésirables associés à des modifications des paramètres biochimiques et hématologiques ont été déterminées d’après la fréquence des anomalies biologiques par rapport à l’inclusion. a Inclut : pneumonie atypique, pneumonie, pneumonie d’inhalation, pneumonie à cryptocoque, infection des voies respiratoires inférieures, infection virale des voies respiratoires inférieures, infection pulmonaire b Inclut les événements de grade 5 c Grade non applicable d Inclut : céphalée, céphalée d’origine sinusale, gêne au niveau de la tête, migraine, céphalée de tension e Inclut : paresthésies, neuropathie sensorielle périphérique, dysesthésie, hyperesthésie, hypoesthésie, névralgie, neuropathie périphérique, neurotoxicité, neuropathie motrice périphérique, polyneuropathie, sensation de brûlure, névralgie post- herpétique f Inclut : perception visuelle de la profondeur altérée, cataracte, daltonisme acquis, diplopie, glaucome, augmentation de la pression intra-oculaire, œdème maculaire, photophobie, photopsie, œdème rétinien, vision trouble, baisse de l’acuité visuelle, défaut du champ visuel, décollement du vitré, corps flottants vitréens, amaurose fugace g Inclut : bradycardie, bradycardie sinusale h Inclut : tachycardie sinusale, tachycardie, tachycardie auriculaire, augmentation de la fréquence cardiaque i Inclut : augmentation de la pression artérielle, hypertension diastolique, hypertension, hypertension systolique j Inclut : dyspnée, dyspnée d’effort k Inclut : pneumopathie interstitielle diffuse, pneumopathie inflammatoire l Inclut : gêne abdominale, distension abdominale, douleur abdominale, douleur abdominale basse, douleur abdominale haute, inconfort épigastrique m Inclut : stomatite aphteuse, stomatite, ulcère aphteux, ulcération buccale, vésicules buccales n Inclut : dermatite acnéiforme, érythème, éruption cutanée exfoliative, éruption cutanée, éruption cutanée érythémateuse, éruption cutanée maculaire, éruption cutanée maculo-papulaire, éruption cutanée papulaire, éruption cutanée pruritique, éruption cutanée pustulaire, dermatite, dermatite allergique, dermatite de contact, érythème généralisé, éruption cutanée folliculaire, urticaire, éruption médicamenteuse, éruption cutanée toxique o Inclut : prurit, prurit allergique, prurit généralisé, prurit génital, prurit vulvo-vaginal p Inclut : douleurs musculo-squelettiques, myalgie, spasmes musculaires, rigidité musculaire, contractions musculaires, gêne musculo-squelettique q Inclut : asthénie, fatigue r Inclut : œdème de la paupière, œdème de la face, œdème périphérique, œdème péri-orbitaire, gonflement du visage, œdème généralisé, gonflement périphérique, angio-œdème, gonflement des lèvres, gonflement périorbitaire, gonflement de la peau, gonflement des paupières s Inclut : augmentation du cholestérol sanguin, hypercholestérolémie |
|||
Description de certains effets indésirables
Effets indésirables pulmonaires
Dans l’étude ALTA 1L, 2,9 % des patients ont présenté une PID/pneumopathie inflammatoire de tout grade au début du traitement (dans les 8 jours), notamment une PID/pneumopathie inflammatoire de grade 3‑4 chez 2,2 % des patients. Il n’y a eu aucun cas fatal de PID/pneumopathie inflammatoire. De plus, 3,7 % des patients ont présenté une pneumopathie inflammatoire ultérieurement au cours du traitement.
Dans l’étude ALTA, 6,4 % des patients ont présenté des effets indésirables pulmonaires de tous grades, notamment PID/pneumopathie inflammatoire, pneumonie et dyspnée, au début du traitement (dans les 9 jours, délai d’apparition médian : 2 jours) ; 2,7 % des patients ont développé des effets indésirables pulmonaires de grade 3-4 et un patient (0,5 %) a développé une pneumonie d’évolution
fatale. Face à l’apparition d’effets indésirables pulmonaires de grade 1-2, le traitement par Alunbrig a été interrompu et repris ou la posologie d’Alunbrig a été diminuée. Des effets indésirables pulmonaires précoces sont également apparus dans une étude d’escalade de doses chez certains patients (N = 137) (étude 101), y compris trois cas fatals (hypoxie, syndrome de détresse respiratoire aigu et pneumonie).
Par ailleurs, 2,3 % des patients de l’étude ALTA ont présenté une pneumopathie inflammatoire ultérieurement au cours du traitement, dont 2 cas de pneumopathie inflammatoire de grade 3 (voir rubriques Posologie et mode d’administration et Mises en garde spéciales et précautions d’emploi).
Patients âgés
Des effets indésirables pulmonaires précoces ont été rapportés chez 10,1 % des patients dont l’âge
était ≥ 65 ans, contre 3,1 % des patients dont l’âge était < 65 ans.
Hypertension
Une hypertension a été décrite chez 30 % des patients recevant un schéma posologique de 180 mg d’Alunbrig, dont 11 %, une hypertension de grade 3. La posologie a été réduite chez 1,5 % des patients recevant un schéma posologique de 180 mg, du fait de l’hypertension. Les pressions systolique et diastolique moyennes ont augmenté au fil du temps chez tous les patients (voir rubriques Posologie et mode d’administration et Mises en garde spéciales et précautions d’emploi).
Bradycardie
Une bradycardie a été décrite chez 8,4 % des patients recevant un schéma posologique de 180 mg d’Alunbrig.
Des fréquences cardiaques inférieures à 50 battements par minute (bpm) ont été décrites chez 8,4 % des patients recevant un schéma posologique de 180 mg (voir rubriques Posologie et mode d’administration et Mises en garde spéciales et précautions d’emploi)).
Troubles visuels
Des troubles visuels ont été rapportés chez 14 % des patients recevant un schéma posologique de 180 mg d’Alunbrig, dont trois effets indésirables de grade 3 (1,1 %), incluant œdème maculaire et cataracte.
La posologie a été diminuée chez deux patients (0,7 %) recevant un schéma posologique de 180 mg, du fait de troubles visuels (voir rubriques Posologie et mode d’administration et Mises en garde spéciales et précautions d’emploi)).
Neuropathie périphérique
Vingt pour cent des patients recevant un schéma posologique de 180 mg d’Alunbrig ont présenté une neuropathie périphérique. Les effets indésirables de type neuropathie se sont résolus chez 33 % des patients. La durée médiane de la neuropathie périphérique était de 6,6 mois, avec un maximum de 28,9 mois.
Augmentation du taux de la créatine phosphokinase (CPK)
Dans les études ALTA 1L et ALTA, des augmentations du taux de la CPK ont été décrites chez 64 % des patients recevant un schéma posologique de 180 mg d’Alunbrig. L’incidence des augmentations du taux de la CPK de grade 3-4 était de 18 %. Le délai d’apparition médian des augmentations du taux de la CPK était de 28 jours.
La posologie a été diminuée chez 10 % des patients recevant un schéma posologique de 180 mg du fait d’une augmentation du taux de la CPK (voir rubriques Posologie et mode d’administration et Mises en garde spéciales et précautions d’emploi)).
Augmentations des enzymes pancréatiques
Des augmentations des taux de l’amylase et de la lipase ont été décrites chez respectivement 47 % et 54 % des patients recevant un schéma posologique de 180 mg d’Alunbrig. Pour les élévations de grades 3 et 4, les incidences pour l’amylase et la lipase étaient de 7,7 % et 15 %, respectivement. Le délai d’apparition médian des augmentations des taux de l’amylase et de la lipase était respectivement de 17 jours et 29 jours.
La posologie a été diminuée chez respectivement 4,7 % et 2,9 % des patients recevant un schéma posologique de 180 mg d’Alunbrig du fait d’une augmentation des taux de la lipase et de l’amylase (voir rubriques Posologie et mode d’administration et Mises en garde spéciales et précautions d’emploi)).
Hépatotoxicité
Des augmentations des taux d’ALAT et d’ASAT ont été décrites chez respectivement 49 % et 68 % des patients recevant un schéma posologique de 180 mg d’Alunbrig. Pour les élévations de grades 3 et 4, les incidences des augmentations de l’ALAT et l’ASAT étaient de 4,7 % et 3,6 %, respectivement.
La dose a été réduite en raison d’une augmentation du taux d’ALAT et d’ASAT chez respectivement 0,7 % et 1,1 % des patients qui recevaient le schéma posologique de 180 mg (voir rubriques Posologie et mode d’administration et Mises en garde spéciales et précautions d’emploi).
Hyperglycémie
Une hyperglycémie a été décrite chez 61 % des patients. Une hyperglycémie de grade 3 a été décrite chez 6,6 % des patients.
Aucune modification posologique
n’a été effectuée en raison d’une hyperglycémie.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration – voir Annexe V.
SURVEILLANCE :
- Bilan de la fonction hépatique (ASAT, ALAT, Bilirubine totale) : avant le début du traitement, puis toutes les 2 semaines pendant les 3 premiers mois de traitement.
- Mesure de la glycémie (à jeun) : avant le début du traitement puis régulièrement pendant le traitement.
- Les patients doivent faire l'objet d'une surveillance clinique régulière pour détecter l'apparition de nouveaux symptômes respiratoires ou une aggravation de ces symptômes notamment pendant la première semaine de traitement.
- Pression artérielle et fréquence cardiaque : contrôle régulier.
- Taux de CPK, lipase et amylase doivent être mesurées régulièrement pendant le traitement.
PREVENIR IMMEDIATEMENT LE MEDECIN en cas de :
- Maux de tête, sensations vertigineuses, douleurs dans la poitrine ou essouflement, car ils peuvent être révélateurs d’un problème de pression artérielle.
- Troubles visuels tels qu’une sensibilité à la lumière, une vision floue ou l'apparition de flash lumineux.
- Douleur ou sensibilité douloureuse ou une faiblesse au niveau musculaire.
- Douleurs abdominales hautes, y compris des douleurs abdominales s'aggravant avec la consommation d'aliments et pouvant irradier dans le dos, ainsi qu'une perte de poids ou des nausées ,car elles peuvent être révélatrices d'un problème au niveau du pancréas.
- Douleurs dans le ventre du côté droit de l'estomac, coloration jaune de la peau ou du blanc des yeux, urines sombres, car ils peuvent être révélateurs d'un problème touchant le foie.
- Sensation de faim ou de soif plus importante qu’en temps normal, besoin plus fréquent qu’en temps normal d’uriner, fatigue, confusion, car ils peuvent être révélateurs d’un déséquilibre du taux de sucre dans le sang.
- Nouveaux symptômes ou aggravation des symptômes pulmonaires ou respiratoires tels que des douleurs dans la poitrine, de la toux et de la fièvre, car ils peuvent être révélateurs d'un problème pulmonaire.
- Douleurs ou gêne dans la poitrine, modification du rythme cardiaque (rapide ou lent), étoursissements.
Les femmes en âge de procréer doivent UTILISER UNE METHODE EFFICACE DE CONTRACEPTION (non hormonale) pendant toute la durée du traitement et pendant au moins 4 mois après la dernière prise.
Les hommes dont les partenaires féminines sont en âge de procréer DOIVENT UTILISER UNE METHODE DE CONTRACEPTION pendant toute la durée du traitement et la poursuivre pendant au moins 3 mois après l’arrêt du traitement.
EVITER de consommer du pamplemousse ou du jus de pamplemousse pendant le traitement.
NE PAS PRENDRE de préparations à base de plantes contenant du millepertuis (Hypericum perforatum).
PRUDENCE en cas de conduite de véhicules ou d'utilisation de machines (fatigue, troubles visuels, sensations vertigineuses).
Femmes en âge de procréer / Contraception chez l'homme et la femme
Les femmes en âge de procréer, traitées par Alunbrig ne doivent pas débuter une grossesse et les hommes traités par Alunbrig, ne doivent pas concevoir d'enfant pendant le traitement. Les femmes en âge de procréer doivent avoir recours à une méthode efficace de contraception non hormonale pendant toute la durée du traitement par Alunbrig et la poursuivre pendant au moins 4 mois après la dernière dose. Les hommes dont les partenaires féminines sont en âge de procréer doivent utiliser une méthode efficace de contraception pendant toute la durée du traitement par Alunbrig et la poursuivre pendant au moins 3 mois après la dernière dose d'Alunbrig.
Grossesse
Alunbrig peut entraîner des malformations fœtales s'il est administré à une femme enceinte. Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique Données de sécurité précliniques). Il n'existe pas de données cliniques sur l'utilisation d'Alunbrig chez la femme enceinte. Alunbrig ne doit pas être utilisé pendant la grossesse à moins que la situation clinique de la mère ne justifie le traitement. Si la patiente est enceinte ou débute une grossesse pendant l'utilisation d'Alunbrig, elle devra être informée des risques potentiels pour le fœtus.
Allaitement
On ne sait pas si Alunbrig est excrété dans le lait maternel. Les données disponibles ne permettent pas d'exclure une excrétion potentiel dans le lait maternel. L'allaitement doit être interrompu pendant le traitement par Alunbrig.
Fertilité
Aucune donnée sur l'effet d'Alunbrig sur la fertilité humaine n'est disponible. D'après des études de toxicité à doses répétées menées sur des animaux mâles, Alunbrig peut provoquer une baisse de la fertilité chez les mâles (voir rubrique Données de sécurité précliniques). La pertinence clinique de ces données en matière de fertilité humaine n'est pas connue.
Agents susceptibles d'augmenter les concentrations plasmatiques de brigatinib
Inhibiteurs du CYP3A
Des études in vitro ont démontré que le brigatinib est un substrat du CYP3A4/5. Chez les sujets sains, l'administration concomitante de plusieurs doses biquotidiennes de 200 mg d'itraconazole, un puissant inhibiteur du CYP3A, et d'une dose unique de 90 mg de brigatinib a augmenté la Cmax de brigatinib de 21 %, l'ASC0-INF de 101 % (facteur de 2) et l'ASC0-120 de 82 % (facteur < 2), par rapport à l'administration d'une dose de 90 mg de brigatinib seule. L'utilisation concomitante d'inhibiteurs puissants du CYP3A et d'Alunbrig, y compris notamment certains antiviraux (par ex. indinavir, nelfinavir, ritonavir, saquinavir), des antibiotiques macrolides (par ex. clarithromycine, télithromycine, troléandomycine), des antifongiques (par ex. kétoconazole, voriconazole), et la néfazodone doit être évitée. Si l'utilisation concomitante d'inhibiteurs puissants du CYP3A ne peut être évitée, la dose d'Alunbrig doit être réduite d'environ 50 % (c.-à-d. de 180 mg à 90 mg ou de 90 mg à 60 mg). Après l'arrêt d'un inhibiteur puissant du CYP3A, le traitement par Alunbrig doit être repris au palier de dose toléré avant l'initiation du traitement par inhibiteur puissant du CYP3A.
Les inhibiteurs modérés du CYP3A (par ex. diltiazem et vérapamil) sont susceptibles d'augmenter l'ASC du brigatinib d'environ 40 % d'après des simulations réalisées à partir d'un modèle pharmacocinétique physiologique. Aucune adaptation de la posologie d'Alunbrig n'est nécessaire en cas d'administration concomitante avec des inhibiteurs modérés du CYP3A. Les patients doivent être rigoureusement surveillés quand Alunbrig est co-administré avec des inhibiteurs modérés du CYP3A.
Le pamplemousse ou le jus de pamplemousse peut également augmenter les concentrations plasmatiques de brigatinib et doit être évité (voir rubrique Posologie et mode d'administration).
Inhibiteurs du CYP2C8
Des études in vitro ont démontré que le brigatinib est un substrat du CYP2C8. Chez les sujets sains, l'administration concomitante de plusieurs doses biquotidiennes de 600 mg de gemfibrozil, un puissant inhibiteur du CYP2C8, et d'une dose unique de 90 mg de brigatinib a diminué la Cmax de brigatinib de 41 %, l'ASC0-INF de 12 % et l'ASC0-120 de 15 %, par rapport à l'administration d'une dose de 90 mg de brigatinib seule. L'effet du gemfibrozil sur la pharmacocinétique du brigatinib n'est pas cliniquement significatif et le mécanisme sous-jacent de la diminution de l'exposition du brigatinib n'est pas connu. Aucune adaptation de la posologie n'est nécessaire en cas de co-administration avec de puissants inhibiteurs du CYP2C8.
Inhibiteurs de la P-gp et de la BCRP
Le brigatinib est un substrat de la P-glycoprotéine (P-gp) et de la protéine de résistance du cancer du sein (Breast Cancer Resistance Protein, BCRP) in vitro. Comme le brigatinib présente une solubilité et une perméabilité élevées, l'inhibition de la P-gp et de la BCRP ne devrait pas induire un changement cliniquement significatif de l'exposition systémique au brigatinib. Aucune adaptation de la posologie d'Alunbrig n'est nécessaire en cas d'administration concomitante avec des inhibiteurs de la P-gp et de la BCRP.
Agents susceptibles de diminuer les concentrations plasmatiques de brigatinib
Inducteurs du CYP3A
Chez les sujets sains, l'administration concomitante de plusieurs doses de 600 mg par jour de rifampicine, un puissant inducteur du CYP3A, et d'une dose unique de 180 mg de brigatinib a diminué la Cmax de brigatinib de 60 %, l'ASC0-INF de 80 % (facteur de 5) et l'ASC0-120 de 80 % (facteur de 5), par rapport à l'administration d'une dose de 180 mg de brigatinib seule. L'utilisation concomitante de puissants inducteurs du CYP3A et d'Alunbrig, y compris notamment la rifampicine, la carbamazépine, la phénytoïne, la rifabutine, le phénobarbital et le millepertuis, doit être évitée.
Les inducteurs modérés du CYP3A sont susceptibles de diminuer l'ASC du brigatinib d'environ 50 % d'après des simulations réalisées à partir d'un modèle pharmacocinétique physiologique. L'utilisation concomitante d'inducteurs modérés du CYP3A et d'Alunbrig, y compris notamment l'éfavirenz, le modafinil, le bosentan, l'étravirine et la nafcilline, doit être évitée.
Agents susceptibles de modifier les concentrations plasmatiques de brigatinib
Substrats du CYP3A
Des études in vitro réalisées sur des hépatocytes ont montré que le brigatinib est un inducteur du CYP3A4. Aucune étude d'interaction médicamenteuse avec des substrats sensibles au CYP3A n'a été menée. Le brigatinib est susceptible de diminuer les concentrations plasmatiques des médicaments co- administrés qui sont principalement métabolisés par le CYP3A. Par conséquent, la co-administration d'Alunbrig et de substrats du CYP3A ayant un indice thérapeutique étroit (par ex. alfentanil, fentanyl, quinidine, ciclosporine, sirolimus, tacrolimus) doit être évitée au risque que leur efficacité soit diminuée.
Alunbrig est également susceptible d'induire d'autres enzymes et transporteurs (par ex. CYP2C, P-gp) via les mêmes mécanismes responsables de l'induction du CYP3A (par ex. activation du récepteur Pregnane X Receptor).
Substrats transporteurs
La co-administration de brigatinib et de substrats de la P-gp (par ex. digoxine, dabigatran, colchicine, pravastatine), de la BCRP (par ex. méthotrexate, rosuvastatine, sulfasalazine), du transporteur de cations organiques 1 (OCT1), des protéines d'extrusion de multimédicaments et toxines 1 (MATE1) et de 2K (MATE2K) est susceptible d'augmenter leurs concentrations plasmatiques. Les patients doivent être surveillés rigoureusement lorsqu'Alunbrig est co-administré avec des substrats de ces transporteurs ayant un indice thérapeutique étroit (par ex. digoxine, dabigatran, méthotrexate).
Le traitement par Alunbrig doit être initié et supervisé par un médecin expérimenté dans l'utilisation de médicaments anticancéreux.
Le statut du CBNPC ALK-positif doit être connu avant l'initiation du traitement par Alunbrig. Un test validé ALK est nécessaire pour sélectionner les patients présentant un CBNPC ALK-positif (voir rubrique Propriétés pharmacodynamiques). L'évaluation du CBNPC ALK-positif doit être réalisée dans un laboratoire disposant de la technologie spécifique nécessaire.
Posologie
La dose initiale recommandée d'Alunbrig est de 90 mg une fois par jour pendant les 7 premiers jours, puis de 180 mg une fois par jour.
En cas d'interruption du traitement par Alunbrig pendant 14 jours ou plus pour des raisons autres que la survenue d'effets indésirables, le traitement devra être réduit à 90 mg une fois par jour pendant
7 jours avant d'augmenter la dose jusqu'à celle précédemment tolérée.
En cas d'oubli d'une prise ou de vomissements survenant après la prise d'une dose, aucune autre dose ne doit être administrée et la dose suivante doit être prise à l'heure habituelle.
Le traitement doit être poursuivi tant qu'un bénéfice clinique est observé.
Adaptations de dose
Une interruption du traitement et/ou une réduction de dose peut/peuvent être nécessaire(s) en fonction de la tolérance individuelle.
Les niveaux de réduction de la dose d'Alunbrig sont résumés dans le tableau 1.
Tableau 1 : Paliers de réduction de la dose d'Alunbrig recommandés
| Dose | Paliers de dose | ||
| Premier | Deuxième | Troisième | |
| 90 mg une fois par jour (7 premiers jours) | réduire à 60 mg une fois par jour | arrêt définitif | sans objet |
| 180 mg une fois par jour | réduire à 120 mg une fois par jour | réduire à 90 mg une fois par jour | réduire à 60 mg une fois par jour |
Le traitement par Alunbrig doit être arrêté définitivement si le patient ne tolère pas la dose de 60 mg une fois par jour.
Les recommandations de modifications de la posologie d'Alunbrig en cas de survenue d'effets indésirables sont résumées dans le tableau 2.
Tableau 2 : Recommandations d'adaptation de la posologie d'Alunbrig en cas d'effets indésirables
|
Effet indésirable |
Sévérité* |
Adaptation posologique |
|
Pneumopathie interstitielle diffuse (PID)/pneumopathie inflammatoire |
Grade 1 |
· Si l'événement survient pendant les 7 premiers jours de traitement, suspendre le traitement par Alunbrig jusqu'à la récupération de l'état initial puis reprendre au même palier de dose et ne pas augmenter à la dose de 180 mg une fois par jour. · Si la PID/pneumopathie inflammatoire survient après les 7 premiers jours de traitement, suspendre le traitement par Alunbrig jusqu'à la récupération de l'état initial puis reprendre au même palier de dose. · Arrêt définitif du traitement par Alunbrig en cas de récidive de PID/pneumopathie inflammatoire. |
|
Grade 2 |
· Si la PID/pneumopathie inflammatoire survient pendant les 7 premiers jours de traitement, suspendre le traitement jusqu'à la récupération de l'état initial puis reprendre au palier de dose inférieur (voir tableau 1) et ne pas augmenter à la dose de 180 mg une fois par jour. · Si la PID/pneumopathie inflammatoire survient après les 7 premiers jours de traitement, suspendre le traitement par Alunbrig jusqu'à la récupération de l'état initial puis reprendre le traitement par Alunbrig au palier de dose inférieur (voir tableau 1). · Arrêt définitif du traitement par Alunbrig en cas de récidive de PID/pneumopathie inflammatoire. | |
|
Grade 3 ou 4 |
· Arrêt définitif du traitement par Alunbrig. | |
|
Hypertension |
Hypertension de grade 3 (PAS ≥ 160 mmHg ou PAD ≥ 100 mmHg, intervention médicale indiquée, nécessitant plus d'un médicament antihypertenseur ou un traitement plus intensif que celui précédemment utilisé) |
· Le traitement par Alunbrig doit être suspendu jusqu'à récupération d'une hypertension de grade ≤ 1 (PAS < 140 mmHg et PAD < 90 mmHg), puis repris à la même posologie. · En cas de récidive de l'hypertension de grade 3, le traitement par Alunbrig doit être suspendu jusqu'à récupération d'une hypertension de grade ≤ 1, puis repris au palier de dose inférieur (voir tableau 1) ou arrêté définitivement. |
|
Hypertension de grade 4 (conséquences menaçant le pronostic vital, indication d'une intervention en urgence) |
· Le traitement par Alunbrig doit être suspendu jusqu'à récupération d'une hypertension de grade ≤ 1 (PAS < 140 mmHg et PAD < 90 mmHg), puis repris au palier de dose inférieur (voir tableau 1) ou arrêté définitivement. · Arrêt définitif du traitement par Alunbrig en cas de récidive de l'hypertension de grade 4. |
|
Effet indésirable |
Sévérité* |
Adaptation posologique |
|
Bradycardie (fréquence cardiaque inférieure à 60 bpm) |
Bradycardie symptomatique |
· Le traitement par Alunbrig doit être suspendu jusqu'à récupération d'une bradycardie asymptomatique ou une fréquence cardiaque au repos de 60 bpm ou plus. · Si un traitement concomitant pouvant entraîner une bradycardie est identifié et interrompu, ou si la posologie de ce traitement est ajustée, reprendre le traitement par Alunbrig à la même posologie dès la récupération d'une bradycardie asymptomatique ou d'une fréquence cardiaque au repos de 60 bpm ou plus. · Si aucun traitement concomitant pouvant entraîner une bradycardie n'est identifié, ou si ce traitement concomitant ne peut être interrompu ou ajusté, le traitement par Alunbrig doit être repris au palier de dose inférieur (voir tableau 1) dès la récupération d'une bradycardie asymptomatique ou d'une fréquence cardiaque au repos de 60 bpm ou plus. |
|
Bradycardie avec conséquences menaçant le pronostic vital, indication d'une intervention en urgence |
· Si un traitement concomitant pouvant entraîner cet événement est identifié et arrêté, ou si sa posologie est ajustée, le traitement par Alunbrig doit être repris au palier de dose inférieur (voir tableau 1) dès la récupération d'une bradycardie asymptomatique ou d'une fréquence cardiaque au repos de 60 bpm ou plus, avec une surveillance fréquente comme cliniquement indiqué. · Arrêt définitif du traitement par Alunbrig, si aucun médicament concomitant pouvant entraîner cet événement n'est identifié. · Arrêt définitif du traitement par Alunbrig en cas de récidive. | |
|
Augmentation du taux de CPK |
Augmentation de grade 3 ou 4 du taux de CPK (> 5,0 × LSN) avec douleur ou faiblesse musculaire de grade ≥ 2 |
· Le traitement par Alunbrig doit être suspendu jusqu'à la récupération d'un taux de CPK de grade ≤ 1 (≤ 2,5 × LSN) ou aux valeurs initiales, puis repris à la même posologie. · En cas de récidive d'un grade 3 ou 4 avec une douleur ou une faiblesse musculaire de grade ≥ 2, le traitement par Alunbrig doit être suspendu jusqu'à la récupération d'un taux de CPK de grade ≤ 1 (≤ 2,5 × LSN) ou aux valeurs initiales, puis repris au palier de dose inférieur (voir tableau 1). |
|
Augmentation du taux de lipase ou d'amylase |
Augmentation de grade 3 du taux de lipase ou d'amylase (> 2,0 × LSN) |
· Le traitement par Alunbrig doit être suspendu jusqu'à la récupération d'un grade ≤ 1 (≤ 1,5 × LSN) ou aux valeurs initiales, puis repris à la même posologie · En cas de récidive, le traitement par Alunbrig doit être suspendu jusqu'à la récupération d'un grade ≤ 1 (≤ 1,5 × LSN) ou aux valeurs initiales, puis repris au palier de dose inférieur (voir tableau 1). |
|
Effet indésirable |
Sévérité* |
Adaptation posologique |
|
|
Augmentation de grade 4 du taux de lipase ou d'amylase (> 5,0 × LSN) |
· Le traitement par Alunbrig doit être suspendu jusqu'à la récupération d'un grade ≤ 1 (≤ 1,5 × LSN), puis repris au palier de dose inférieur (voir tableau 1). |
|
Hépatotoxicité |
Augmentation de grade ≥ 3 (> 5,0 × LSN) de l'alanine aminotransférase (ALAT) ou de l'aspartate aminotransférase (ASAT) avec bilirubine £ 2 × LSN |
· Le traitement par Alunbrig doit être suspendu jusqu'à la récupération des valeurs initiales ou inférieures ou égales à 3 × LSN, puis repris au palier de dose inférieur (voir tableau 1). |
|
Augmentation de grade ≥ 2 (> 3 × LSN) de l'ALAT ou de l'ASAT et de la bilirubine totale > 2 × LSN en l'absence de cholestase ou d'hémolyse |
· Arrêt définitif du traitement par Alunbrig. | |
|
Hyperglycémie |
Grade 3 (supérieure à 250 mg/dL ou 13,9 mmol/L) ou supérieur |
· En l'absence d'un contrôle glycémique adéquat avec une prise en charge médicale adaptée, le traitement par Alunbrig doit être suspendu jusqu'à obtention d'un contrôle glycémique satisfaisant. Dès obtention d'un équilibre glycémique satisfaisant, le traitement par Alunbrig peut être repris au palier de dose inférieur (voir tableau 1) ou arrêté définitivement. |
|
Troubles visuels |
Grade 2 ou 3 |
· Le traitement par Alunbrig doit être suspendu jusqu'à la récupération d'un grade 1 ou aux valeurs initiales, puis repris au palier de dose inférieur (voir tableau 1). |
|
Grade 4 |
· Arrêt définitif du traitement par Alunbrig. | |
|
Autres effets indésirables |
Grade 3 |
· Le traitement par Alunbrig doit être suspendu jusqu'à la récupération des valeurs initiales, puis repris à la même posologie. · En cas de récidive, le traitement par Alunbrig doit être suspendu jusqu'à la récupération des valeurs initiales, puis repris au palier de dose inférieur (voir tableau 1) ou arrêté définitivement. |
|
Grade 4 |
· Le traitement par Alunbrig doit être suspendu jusqu'à la récupération des valeurs initiales, puis repris au palier de dose inférieur (voir tableau 1). · En cas de récidive le traitement par Alunbrig doit être suspendu jusqu'à la récupération des valeurs initiales, puis repris au palier de dose inférieur (voir tableau 1) ou arrêté définitivement. | |
|
bpm = battements par minute ; CPK = créatine phosphokinase ; PAD = pression artérielle diastolique ; PAS = pression artérielle systolique ; LSN = limite supérieure de la normale | ||
Populations particulières
Patients âgés
Les données limitées sur la sécurité et l'efficacité d'Alunbrig chez les patients âgés de 65 ans et plus suggèrent qu'aucune adaptation posologique n'est nécessaire chez les patients âgés (voir rubrique Effets indésirables). Aucune donnée n'est disponible sur les patients de plus de 85 ans.
Insuffisance hépatique
Aucune adaptation posologique d'Alunbrig n'est nécessaire pour les patients présentant une insuffisance hépatique légère (score de Child-Pugh A) ou modérée (score de Child-Pugh B). Une dose initiale réduite de 60 mg une fois par jour pendant les 7 premiers jours suivie d'une dose de 120 mg par jour est recommandée pour les patients présentant une insuffisance hépatique sévère (score de Child-Pugh C) (voir rubrique Propriétés pharmacocinétiques).
Insuffisance rénale
Aucune adaptation posologique d'Alunbrig n'est nécessaire pour les patients présentant une insuffisance rénale légère ou modérée (débit de filtration glomérulaire estimé (DFGe) ≥ 30 mL/min). Une dose initiale réduite de 60 mg une fois par jour pendant les 7 premiers jours suivie d'une dose de 90 mg par jour est recommandée pour les patients présentant une insuffisance rénale sévère
(DFGe < 30 mL/min) (voir rubrique Propriétés pharmacocinétiques). Les patients présentant une insuffisance rénale sévère doivent faire l'objet d'une surveillance étroite afin de détecter l'apparition ou l'aggravation de symptômes respiratoires pouvant évoquer une PID/pneumopathie inflammatoire (par ex. dyspnée, toux, etc.), en particulier au cours de la première semaine (voir rubrique Mises en garde et précautions d'emploi).
Population pédiatrique
La sécurité et l'efficacité d'Alunbrig chez les patients de moins de 18 ans n'ont pas encore été établies. Aucune donnée n'est disponible.
Mode d'administration
Alunbrig est administré par voie orale. Les comprimés doivent être avalés en entier avec de l'eau. Alunbrig peut être pris pendant ou en dehors des repas.
Le pamplemousse ou le jus de pamplemousse peuvent augmenter les concentrations plasmatiques de brigatinib et doivent être évités (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Durée de conservation :
3 ans
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
Il n'y a pas d'antidote spécifique en cas de surdosage avec Alunbrig. En cas de surdosage, surveillez le patient afin de déceler tout effet indésirable (voir rubrique Effets indésirables) et une prise en charge appropriée doit être mise en place.
Classe pharmacothérapeutique : agent antinéoplasique, inhibiteurs de protéine kinase, code ATC : L01XE43
Mécanisme d'action
Le brigatinib est un inhibiteur de la tyrosine kinase ciblant ALK, ROS1 (c-ros oncogene 1) et IGF-1R (insulin-like growth factor 1 receptor). Le brigatinib a inhibé l'autophosphorylation d'ALK et la phosphorylation médiée par ALK de la protéine de signalisation située en aval STAT3 dans les études in vitro et in vivo.
Le brigatinib a inhibé la prolifération in vitro des lignées cellulaires exprimant les protéines de fusion EML4-ALK et NPM-ALK et a démontré une inhibition dose-dépendante de la croissance de xénogreffe de CBNPC EML4-ALK-positif chez la souris. Le brigatinib a inhibé la viabilité in vitro et in vivo des cellules exprimant les formes mutantes d'EML4-ALK associées à une résistance aux inhibiteurs d'ALK, y compris G1202R et L1196M.
Électrophysiologie cardiaque
Dans l'étude 101, le potentiel de prolongation de l'intervalle QT d'Alunbrig a été évalué chez 123 patients présentant des affections malignes avancées après une dose de 30 mg à 240 mg de brigatinib par jour. La modification moyenne maximale de l'intervalle QTcF (intervalle QT corrigé selon la méthode de Fridericia) depuis l'inclusion était inférieure à 10 msec. Une analyse de l'exposition et de l'intervalle QT a montré que la prolongation de l'intervalle QTc n'était pas dépendante de la concentration.
Efficacité et sécurité cliniques
ALTA 1L
La sécurité et l'efficacité d'Alunbrig ont été évaluées dans un essai multicentrique randomisé (selon un rapport 1:1) en ouvert (ALTA 1L) chez 275 patients adultes atteints d'un CBNPC ALK-positif avancé n'ayant pas reçu de traitement antérieur ciblant ALK. Les critères d'éligibilité ont permis l'inclusion de patients présentant un réarrangement d'ALK documenté sur la base d'un test standard local et d'un score de performance ECOG de 0‑2. Les patients pouvaient avoir reçu jusqu'à 1 traitement de chimiothérapie antérieur dans le contexte d'une maladie localement avancée ou métastatique. Les patients stables sur le plan neurologique et présentant des métastases traitées ou non traitées du système nerveux central (SNC), y compris des métastases leptoméningées, étaient éligibles. Les patients ayant des antécédents de maladie pulmonaire interstitielle diffuse, de pneumopathie inflammatoire médicamenteuse ou de pneumopathie radique ont été exclus.
Les patients étaient randomisés selon un rapport 1:1 pour recevoir un traitement par Alunbrig à une dose de 180 mg une fois par jour avec une période initiale de 7 jours à 90 mg une fois par jour (N = 137) ou par crizotinib à 250 mg par voie orale deux fois par jour (N = 138). La randomisation a été stratifiée en fonction des métastases cérébrales (présence, absence) et de l'administration antérieure d'une chimiothérapie dans un contexte de maladie localement avancée ou métastatique (oui, non).
Le principal critère d'évaluation était la survie sans progression (SSP) selon les critères RECIST (Response Evaluation Criteria In Solid Tumours v1.1) évaluée par un comité de revue indépendant en aveugle (Blinded Independent Review Committee, BIRC). Les autres critères d'évaluation évalués par le BIRC incluaient notamment : le taux de réponse objective (TRO) confirmée, la durée de la réponse (DR), le délai avant la réponse, le taux de contrôle de la maladie (TCM), le TRO intracrânienne, la SSP intracrânienne et la DR intracrânienne. Les résultats évalués par l'investigateur comprenaient la SSP et la survie globale.
Les données démographiques et les caractéristiques de la maladie à l'inclusion dans l'essai ALTA 1L étaient : âge médian de 59 ans (intervalle : 27 à 89 ; 32 % de patients âgés d'au moins 65 ans), 59 % d'origine caucasienne et 39 % d'origine asiatique, 55 % de femmes, 39 % des patients avaient un score de performance ECOG de 0 et 56 % un score de performance ECOG de 1, 58 % des patients n'avaient jamais fumé, 93 % des patients étaient atteints d'une maladie de stade IV, 96 % de patients avaient un adénocarcinome, 30 % avaient des métastases du SNC à l'inclusion, 14 % avaient reçu une radiothérapie cérébrale antérieure et 27 % avaient reçu une chimiothérapie antérieure. Les sites de métastases extra‑thoraciques comprenaient le cerveau (30 % des patients), les os (31 % des patients) et le foie (20 % des patients). L'intensité de la dose relative médiane était de 97 % pour Alunbrig et de 99 % pour le crizotinib.
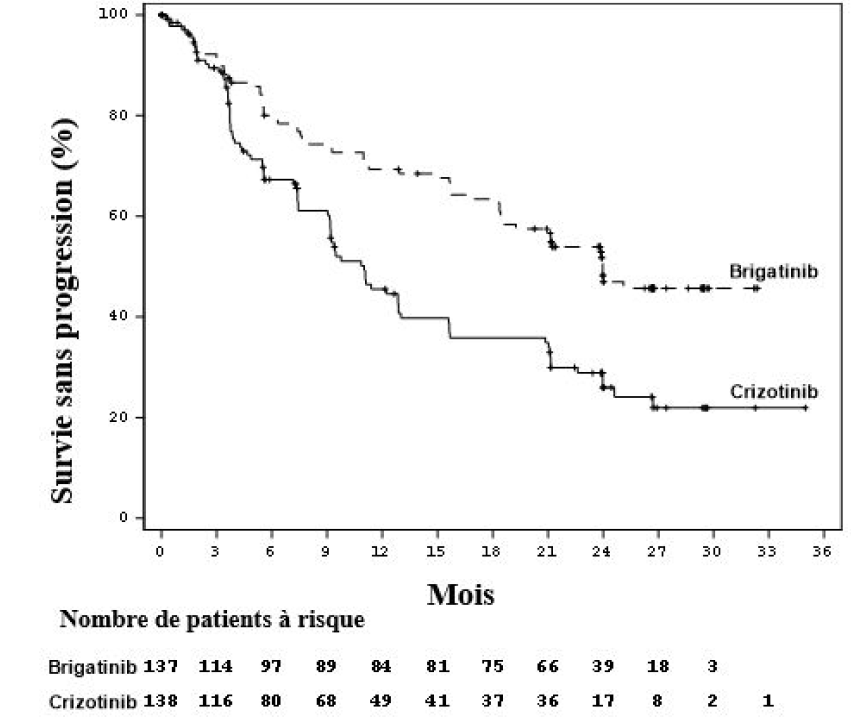
Lors de la première analyse réalisée après une durée médiane de suivi de 11 mois dans le bras Alunbrig, l'étude ALTA 1L a atteint son critère d'évaluation principal démontrant une amélioration statistiquement significative de la SSP évaluée par le BIRC. Les résultats de l'étude reposent sur une analyse de l'efficacité définie par un protocole et réalisée après une durée médiane de suivi de 24,9 mois dans le bras Alunbrig (tableau 4 et figure 1).
Tableau 4 : Résultats d'efficacité dans l'étude ALTA 1L (population ITT)
| Paramètres d'efficacité | Alunbrig N = 137 | Crizotinib N = 138 |
| Durée médiane du suivi (mois) | 24,9 (intervalle : 0-34,1) | 15,2 (intervalle : 0,1-36) |
| Principaux paramètres d'efficacité | ||
| SSP (BIRC) | ||
| Nombre de patients ayant présenté des événements, n (%) | 63 (46 %) | 87 (63 %) |
| Progression de la maladie, n (%) | 56 (40,9 %)a | 82 (59,4 %)b |
| Décès, n (%) | 7 (5,1 %) | 5 (3,6 %) |
| Médiane (en mois) (IC à 95 %) | 24 (18,5 ; NE) | 11 (9,2 ; 12,9) |
| Hazard ratio (IC à 95 %) | 0,49 (0,35 ; 0,68) | |
| Valeur de p selon test log rankc | < 0,0001 | |
| Paramètres d'efficacité secondaires | ||
| Taux de réponse objective confirmée (BIRC) | ||
| Répondeurs, n (%) (IC à 95 %) | 101 (73,7 %) (65,5 ; 80,9) | 85 (61.6 %) (52.9 ; 69.7) |
| Valeur de pc,d | 0,0342 | |
| Réponse complète, % | 14,6 % | 8,7 % |
| Réponse partielle, % | 59,1 % | 52,9 % |
| Durée de réponse confirmée (BIRC) | ||
| Médiane (en mois) (IC à 95 %) | NE (19,4 ; NE) | 13,8 (9,3 ; 20,8) |
| Survie globale | ||
| Nombre d'événements, n (%) | 33 (24,1) | 37 (26,8) |
| Médiane (en mois) (IC à 95 %) | NE (NE ; NE) | NE (NE ; NE) |
| Risque relatif (IC à 95 %) | 0,92 (0,57 ; 1,47) | |
| Valeur de p selon test log rankd | 0,7710 | |
BIRC = Blinded Independent Review Committee (comité de revue indépendant en aveugle) ; NE = non estimable ;
IC = intervalle de confiance
a Inclut 2 patients ayant reçu une radiothérapie cérébrale palliative
b Inclut 8 patients ayant reçu une radiothérapie cérébrale palliative
c Stratifié en fonction de la présence de métastases isolées du SNC à l'inclusion et l'administration antérieure d'une chimiothérapie dans le cadre d'une maladie localement avancée ou métastatique pour le test du log-rank et le test de Cochran-Mantel-Haenszel, respectivement
d D'après un test de Cochran-Mantel-Haenszel
Figure 1 : Courbe de Kaplan-Meier de la survie sans progression évaluée le BIRC dans l'étude ALTA 1L
L'évaluation par le BIRC de l'efficacité intracrânienne selon les critères RECIST v1.1 chez les patients présentant des métastases cérébrales de tout type et les patients présentant des métastases cérébrales mesurables (diamètre le plus long ≥ 10 mm) à l'inclusion est résumée dans le tableau 5.
Tableau 5 : Efficacité intracrânienne évaluée par le BIRC chez des patients de l'étude ALTA 1L
| Paramètres d'efficacité | Patients présentant des métastases cérébrales mesurables à l'inclusion | |
| Alunbrig N = 18 | Crizotinib N = 23 | |
| Taux de réponse objective intracrânienne confirmée | ||
| Répondeurs, n (%) (IC à 95 %) | 14 (77,8 %) (52,4 ; 93,6) | 6 (26,1 %) (10,2 ; 48,4) |
| Valeur de pa,b | 0,0014 | |
| Réponse complète, % | 27,8 % | 0 |
| Réponse partielle, % | 50 % | 26,1 % |
| Durée de la réponse intracrânienne confirméec | ||
| Médiane (mois) (IC à 95 %) | NE (5,7 ; NE) | 9,2 (3,9 ; 9,2) |
| | Patients présentant des métastases cérébrales de tout type à l'inclusion | |
| Alunbrig N = 47 | Crizotinib N = 49 | |
| Taux de réponse objective intracrânienne confirmée | ||
| Répondeurs, n (%) (IC à 95 %) | 31 (66 %) (50,7 ; 79,1) | 8 (16,3 %) (7,32 ; 29,7) |
| Valeur de pa,b | < 0,0001 | |
| Réponse complète, % | 44,7 % | 4,1 % |
| Réponse partielle, % | 21,3 % | 12,2 % |
| Durée de la réponse intracrânienne confirméec | ||
| Médiane (en mois) (IC à 95 %) | 24 (16,9 ; NE) | 9,2 (3,9 ; NE) |
| SSP intracrânienned | | |
| Nombre de patients ayant présenté des événements, n (%) | 21 (44,7 %) | 32 (65,3 %) |
| Progression de la maladie, n (%) | 21 (44,7 %)e | 29 (59,2 %)f |
| Décès, n (%) | 0 | 3 (6,1 %) |
| Médiane (en mois) (IC à 95 %) | 24 (13 ; NE) | 5,6 (3,7 ; 7,5) |
| Hazard ratio (IC à 95 %) | 0,31 (0,17 ; 0,56) | |
| Valeur de p selon test log rank | < 0,0001 | |
IC = intervalle de confiance ; NE = non estimable
a Stratifié en fonction de l'administration antérieure d'une chimiothérapie dans le cadre d'une maladie localement avancée ou métastatique pour le test du log-rank et le test de Cochran-Mantel-Haenszel, respectivement.
b D'après un test de Cochran-Mantel-Haenszel
c Mesurée à partir de la date de la première réponse intracrânienne confirmée jusqu'à la date de progression de la maladie intracrânienne (nouvelles lésions intracrâniennes, augmentation ≥ 20 % du diamètre de la lésion intracrânienne cible par rapport au nadir ou progression non équivoque des lésions intracrâniennes non cibles) ou décès ou censure.
d Mesurée à partir de la date de randomisation jusqu'à la date de progression de la maladie intracrânienne (nouvelles lésions intracrâniennes, augmentation ≥ 20 % du diamètre de la lésion intracrânienne cible par rapport au nadir ou progression non équivoque des lésions intracrâniennes non cibles) ou décès ou censure.
e Inclut 1 patient ayant reçu une radiothérapie cérébrale palliative
f Inclut 2 patients ayant reçu une radiothérapie cérébrale palliative
ALTA
L'efficacité et la sécurité cliniques d'Alunbrig ont été évaluées dans un essai multicentrique randomisé (selon un rapport de 1:1) en ouvert (ALTA) chez 222 patients adultes atteints d'un CBNPC ALK- positif localement avancé ou métastatique, dont la maladie a progressé sous crizotinib. Les critères d'éligibilité ont permis l'inclusion de patients présentant un réarrangement d'ALK documenté sur la base d'un test validé, un score de performance ECOG de 0-2 et des traitements par chimiothérapie antérieurs. Par ailleurs, les patients présentant des métastases du système nerveux central (SNC) étaient inclus, à condition qu'elles soient stables sur le plan neurologique et ne nécessitent pas une augmentation de la dose de corticoïdes. Les patients ayant des antécédents de maladie pulmonaire interstitielle diffuse ou de pneumopathie inflammatoire médicamenteuse ont été exclus.
Les patients étaient randomisés selon un rapport 1:1 pour recevoir un traitement par Alunbrig à une dose de 90 mg une fois par jour (schéma posologique à 90 mg, N = 112) ou à une dose de 180 mg une fois par jour avec une période initiale de 7 jours à 90 mg une fois par jour (schéma posologique à 180 mg, N = 110). La durée médiane du suivi était de 22,9 mois. La randomisation a été stratifiée en fonction des métastases cérébrales (présence, absence) et de la meilleure réponse antérieure au traitement par crizotinib (réponse complète ou partielle, toute autre réponse/inconnue).
Le principal critère d'évaluation était le taux de réponse objective (TRO) selon les critères RECIST (Response Evaluation Criteria In Solid Tumors v1.1) évalué par l'investigateur. Les autres critères d'évaluation incluaient notamment : TRO évalué par un comité de revue indépendant (IRC, Independent Review Committee), le délai avant la réponse, la survie sans progression (SSP), la durée de la réponse (DR), la survie globale, le TRO intracrânienne et la DR intracrânienne, d'après l'évaluation d'un IRC.
Les données démographiques et les caractéristiques de la maladie à l'inclusion dans l'étude ALTA étaient : âge médian de 54 ans (intervalle : 18 à 82 ans ; 23 % de patients âgés d'au moins 65 ans), 67 % d'origine caucasienne et 31 % d'origine asiatique, 57 % de femmes, 36 % de patients avaient un score de performance ECOG de 0, 57 % un score de performance ECOG de 1 et 7 % un score de performance ECOG de 2, 60 % de patients n'avaient jamais fumé, 35 % des patients étaient d'anciens fumeurs et 5 % étaient fumeurs, 98 % des patients étaient au stade IV, 97 % de patients avaient un adénocarcinome et 74 % avaient reçu une chimiothérapie antérieure. Les sites les plus fréquents de métastases extra thoraciques étaient le cerveau (69 %, dont 62 % ayant reçu une radiothérapie cérébrale antérieure), les os (39 %) et le foie (26 %).
Les résultats d'efficacité de l'analyse de l'étude ALTA sont récapitulés dans le tableau 6 et les courbes de Kaplan-Meier (KM) relatives aux évaluations de SSP effectuées par l'investigateur sont présentées dans la figure 2.
Tableau 6 : Résultats d'efficacité dans l'étude ALTA (population ITT)
| Paramètre d'efficacité | Évaluation de l'investigateur | Évaluation de l'IRC | ||
| Schéma posologique à 90 mg* N = 112 | Schéma posologique à 180 mg† N = 110 | Schéma posologique à 90 mg* N = 112 | Schéma posologique à 180 mg† N = 110 | |
| Taux de réponse objective | ||||
| (%) | 46 % | 56 % | 51 % | 56 % |
| IC‡ | (35 ; 57) | (45 ; 67) | (41 ; 61) | (47 ; 66) |
| Délai de réponse | ||||
| Médiane (mois) | 1,8 | 1,9 | 1,8 | 1,9 |
| Durée de la réponse | ||||
| Médiane (mois) | 12,0 | 13,8 | 16,4 | 15,7 |
| IC à 95 % | (9,2 ; 17,7) | (10,2 ; 19,3) | (7,4 ; 24,9) | (12,8 ; 21,8) |
| Survie sans progression | ||||
| Médiane (mois) | 9,2 | 15,6 | 9,2 | 16,7 |
| IC à 95 % | (7,4 ; 11,1) | (11,1 ; 21) | (7,4 ; 12,8) | (11,6 ; 21,4) |
| Survie globale | ||||
| Médiane (mois) | 29,5 | 34,1 | NA | NA |
| IC à 95 % | (18,2 ; NE) | (27,7 ; NE) | NA | NA |
| Probabilité de survie à 12 mois (%) | 70,3 % | 80,1 % | NA | NA |
IC = intervalle de confiance, NE = non estimable, NA = non applicable
*schéma posologique à 90 mg une fois par jour
†schéma posologique à 180 mg une fois par jour avec période initiale de 7 jours à 90 mg une fois par jour
‡L'intervalle de confiance est de 97,5 % pour le TRO évaluée par l'investigateur et de 95 % pour le TRO évaluée par l'IRC
Figure 2 : Survie sans progression systémique évaluée par l'investigateur : Population ITT par bras de traitement (ALTA)
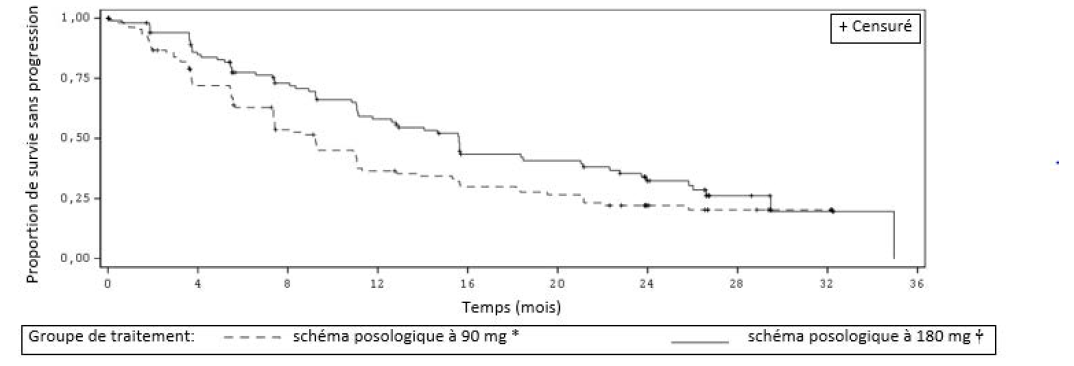
Abréviations : ITT = intention de traiter
Remarque : La survie sans progression était définie comme le temps écoulé entre l'initiation du traitement et la date à laquelle une progression de la maladie ou un décès a été mis(e) en évidence pour la première fois, selon l'événement survenant en premier.
*schéma posologique à 90 mg une fois par jour
†schéma posologique à 180 mg une fois par jour avec période initiale de 7 jours à 90 mg une fois par jour
Les évaluations par l'IRC du TRO intracrânienne et de la durée de la réponse intracrânienne chez les patients de l'étude ALTA présentant des métastases cérébrales mesurables (diamètre en longueur ≥ 10 mm) à l'inclusion sont résumées dans le tableau 7.
Tableau 7 : Efficacité intracrânienne chez les patients présentant des métastases cérébrales mesurables à l'inclusion dans l'étude ALTA
| Paramètre d'efficacité évalué par l'IRC | Patients présentant des métastases cérébrales mesurables à l'inclusion | |
| Schéma posologique à 90 mg* (N = 26) | Schéma posologique à 180 mg† (N = 18) | |
| Taux de réponse objective intracrânienne | ||
| (%) | 50 % | 67 % |
| IC à 95 % | (30 ; 70) | (41 ; 87) |
| Taux de contrôle de la maladie intracrânienne | ||
| (%) | 85 % | 83 % |
| IC à 95 % | (65 ; 96) | (59 ; 96) |
| Durée de la réponse intracrânienne‡, | ||
| Médiane (mois) | 9,4 | 16,6 |
| IC à 95 % | (3,7 ; 24,9) | (3,7 ; NE) |
%IC = intervalle de confiance, NE = non estimable
*Schéma posologique à 90 mg une fois par jour
†Schéma posologique à 180 mg une fois par jour avec période initiale de 7 jours à 90 mg une fois par jour
‡Les événements incluent la progression de la maladie intracrânienne (nouvelles lésions, augmentation du diamètre de la lésion cible intracrânienne ≥ 20 % depuis le nadir, ou progression sans équivoque des lésions non ciblées intracrâniennes) ou décès.
Chez les patients présentant des métastases cérébrales à l'inclusion, le taux de contrôle de la maladie intracrânienne était de 77,8 % (IC à 95 % : 67,2 ;86,3) dans le groupe de traitement ayant reçu le schéma posologique à 90 mg (N = 81) et de 85,1 % (IC à 95 % : 75 ; 92,3) dans le groupe de traitement ayant reçu le schéma posologique à 180 mg (N = 74).
Étude 101
Dans une étude distincte de recherche de dose, 25 patients présentant un CBNPC ALK-positif ayant progressé sous crizotinib ont reçu un traitement par Alunbrig à une dose de 180 mg une fois par jour avec période initiale de 7 jours à 90 mg une fois par jour. Parmi ces patients, 19 ont eu une réponse objective confirmée par l'évaluation de l'investigateur (76 % ; IC à 95 % : 55 ; 91) et la durée de réponse médiane estimée selon Kaplan-Meier parmi les 19 patients répondeurs était de 26,1 mois (IC à 95 % : 7,9 ; 26,1). La SSP médiane estimée selon Kaplan-Meier était de 16,3 mois (IC à 95 % : 9,2 ; NE) et la probabilité de survie globale à 12 mois était de 84,0 % (IC à 95 % : 62,8 ; 93,7).
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec Alunbrig dans tous les sous-groupes de la population pédiatrique présentant un carcinome pulmonaire (carcinome à petites cellules et non à petites cellules) (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Absorption
Dans l'étude 101, après l'administration d'une dose orale unique de brigatinib (30-240 mg) chez des patients, le délai médian d'obtention de la concentration maximale (Tmax) a été de 1-4 heures après l'administration. Après une dose unique et à l'état d'équilibre, l'exposition systémique était proportionnelle à la dose dans l'intervalle de doses de 60-240 mg une fois par jour. Une accumulation modérée a été observée lors de l'administration répétée (moyenne géométrique du rapport d'accumulation : 1,9 à 2,4). La moyenne géométrique de la Cmax à l'état d'équilibre du brigatinib à des doses de 90 mg et de 180 mg une fois par jour a été respectivement de 552 et 1 452 ng/mL, et l'ASC0-t correspondante a été respectivement de 8 165 et 20 276 h·ng/mL. Le brigatinib est un substrat des protéines de transport P-gp et BCRP.
Chez les sujets sains, par rapport à un jeûne d'une nuit, un repas riche en lipides a diminué de 13 % la Cmax du brigatinib sans effet sur l'ASC. Le brigatinib peut être administré pendant ou en dehors des repas.
Distribution
Le brigatinib se liait modérément (91 %) aux protéines plasmatiques humaines et la liaison n'était pas dépendante de la concentration. Le rapport de concentration sang/plasma est de 0,69. Chez les patients ayant reçu un traitement par brigatinib à 180 mg une fois par jour, la moyenne géométrique du volume apparent de distribution (Vz/F) du brigatinib à l'état d'équilibre était de 307 L, indiquant une distribution modérée dans les tissus.
Biotransformation
Les études in vitro ont démontré que le brigatinib est principalement métabolisé par le CYP2C8 et le CYP3A4, et beaucoup moins par le CYP3A5.
Après l'administration orale d'une dose unique de 180 mg de [14C]brigatinib chez des sujets sains, les deux voies majeures d'élimination métabolique étaient la N-déméthylation et la conjugaison à la cystéine. Dans les urines et fécès combinées, respectivement 48 %, 27 % et 9,1 % de la dose radioactive étaient excrétés sous forme de brigatinib inchangée, de N-desméthyl brigatinib (AP26123) et de conjugué de brigatinib à la cystéine. La forme inchangée de brigatinib était le principal composant radioactif circulant (92 %), avec l'AP26123 (3,5 %), le principal métabolite également observé in vitro. Chez les patients, à l'état d'équilibre, l'ASC plasmatique de l'AP26123 était < 10 % de l'exposition au brigatinib. Dans les essais cellulaires et de kinase in vitro, le métabolite AP26123 a inhibé ALK environ 3 fois moins que le brigatinib.
Élimination
Chez les patients ayant reçu un traitement par brigatinib de 180 mg une fois par jour, la moyenne géométrique de la clairance orale apparente (CL/F) du brigatinib à l'état d'équilibre était de 8,9 L/h et la demi-vie d'élimination plasmatique médiane était de 24 h.
Les fécès constituent la principale voie d'excrétion du brigatinib. Chez 6 sujets sains de sexe masculin ayant reçu une dose orale unique de 180 mg de [14C]brigatinib, 65 % de la dose administrée ont été éliminés dans les fécès et 25 % de la dose administrée ont été éliminés dans les urines. La forme inchangée du brigatinib représentait respectivement 41 % et 86 % de la radioactivité totale dans les fécès et les urines, le reste étant des métabolites.
Populations spécifiques
Insuffisance hépatique
La pharmacocinétique du brigatinib a été caractérisée chez des sujets sains présentant une fonction hépatique normale (N = 9), et des patients atteints d'une insuffisance hépatique légère (score de Child-Pugh A, N = 6), des patients atteints d'une insuffisance hépatique modérée (score de Child-Pugh B, N = 6) ou des patients atteints d'une insuffisance hépatique sévère (score de Child-Pugh C, N = 6). La pharmacocinétique du brigatinib était similaire chez les sujets sains présentant une fonction hépatique normale et les patients atteints d'une insuffisance hépatique légère (score de Child-Pugh A) ou modérée (score de Child-Pugh B). L'ASC0-INF du brigatinib non lié était 37 % supérieure chez les patients atteints d'une insuffisance hépatique sévère (score de Child-Pugh C) en comparaison aux sujets sains présentant une fonction hépatique normale (voir rubrique Posologie et mode d'administration).
Insuffisance rénale
La pharmacocinétique du brigatinib est similaire chez les patients présentant une fonction rénale normale et les patients atteints d'une insuffisance rénale légère ou modérée (DFGe ≥ 30 mL/min) d'après les résultats d'analyses pharmacocinétiques de population. Dans une étude de pharmacocinétique, l'ASC0-INF du brigatinib non lié était 94 % supérieure chez les patients atteints d'une insuffisance rénale sévère (DFGe < 30 mL/min, N = 6) en comparaison aux patients présentant une fonction rénale normale (DFGe ≥ 90 mL/min, N = 8) (voir rubrique Posologie et mode d'administration).
Origine ethnique et sexe
Les analyses de pharmacocinétique de population ont montré que l'origine ethnique et le sexe n'avaient aucune incidence sur la pharmacocinétique du brigatinib.
Âge, poids corporel et concentrations d'albumine
Les analyses de pharmacocinétique de population ont montré que le poids corporel, l'âge et les concentrations d'albumine n'avaient aucune répercussion cliniquement significative sur la pharmacocinétique du brigatinib.
Alunbrig a une influence mineure sur l’aptitude àconduire des véicules et àutiliser des machines.
Toutefois, la prudence s'impose lors de la conduite de véicules ou l'utilisation de machines, puisque les patients sont susceptibles de préenter des troubles visuels, des sensations vertigineuses ou une fatigue lors de la prise d’Alunbrig.
Les études de pharmacologie de sécurité évaluant le brigatinib ont identifié un potentiel d'effets pulmonaires (modification de la fréquence respiratoire ; à 1-2 fois la Cmax humaine), d'effets cardiovasculaires (modification de la fréquence cardiaque et de la pression artérielle ; à 0,5 fois la Cmax humaine) et des effets rénaux (diminution de la fonction rénale ; à 1-2,5 fois la Cmax humaine) mais n'ont pas indiqué de potentiel d'allongement de l'intervalle QT ou d'effets neurofonctionnels.
Les effets indésirables observés chez l'animal à des niveaux d'exposition similaires aux niveaux d'exposition clinique avec une possible pertinence pour l'utilisation clinique étaient les suivants : système gastro-intestinal, moelle osseuse, yeux, testicules, foie, reins, os et cœur. Ces effets étaient généralement réversibles pendant la période de récupération sans traitement, cependant les effets oculaires et testiculaires ont été des exceptions en raison d'une absence de récupération.
Dans les études de toxicité à doses répétées, des modifications pulmonaires (macrophages alvéolaires spumeux) ont été observées chez les singes à ≥ 0,2 fois l'ASC humaine ; néanmoins, ces modifications étaient mineures et similaires à celles rapportées initialement chez des singes naïfs de traitement, et aucun signe clinique de détresse respiratoire n'a été constaté chez ces singes.
Aucune étude de carcinogenicité n'a été effectuée avec le brigatinib.
Le brigatinib n'a pas été mutagène in vitro dans le test d'Ames (mutation bactérienne inverse) ou les essais d'aberration chromosomique dans des cellules de mammifères mais a légèrement augmenté le nombre de micronoyaux dans un test de micronoyaux sur des cellules de moelle osseuse de rat. Le mécanisme d'induction de micronoyaux a été une ségrégation chromosomique anormale (aneugénicité) et non un effet clastogène sur les chromosomes. Cet effet a été observé à environ cinq fois l'exposition humaine à la dose de 180 mg une fois par jour.
Le brigatinib est susceptible d'avoir un effet négatif sur la fertilité masculine. Une toxicité testiculaire a été observée dans les études à doses répétées menées chez l'animal. Chez le rat, cette toxicité s'est manifestée par une diminution du poids des testicules, des vésicules séminales et de la prostate, ainsi que par une dégénérescence tubulaire testiculaire ; ces effets n'ont pas été réversibles pendant la période de récupération. Chez le singe, on a observé une diminution de la taille des testicules et des signes d'hypospermatogénèse à l'examen microscopique ; ces effets ont été réversibles pendant la période de récupération. Dans l'ensemble, ces effets sur les organes reproducteurs chez le rat et le singe mâles sont apparus à des expositions ≥ 0,2 fois l'ASC observée chez les patients à la dose de 180 mg une fois par jour. Aucun effet indésirable apparent sur les organes reproducteurs de la femelle n'a été observé dans les études de toxicologie générale effectuées chez le rat et le singe.
Dans une étude de développement embryofœtal dans laquelle des rates gestantes ont reçu des doses quotidiennes de brigatinib pendant l'organogenèse, des anomalies squelettiques liées à la dose ont été observées à des doses aussi faibles qu'environ 0,7 fois l'exposition humaine selon l'ASC à la dose de 180 mg une fois par jour. Ces anomalies se sont manifestées par une létalité embryonnaire, une diminution de la croissance fœtale et des variations squelettiques.
Les patients doivent veiller à conserver la capsule de dessiccant du flacon et à ne pas l'avaler.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I
médicament nécessitant une surveillance particulière pendant le traitement
prescription hospitalière
prescription réservée aux spécialistes et services CANCEROLOGIE
prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE
Comprimé pelliculé ovale blanc à blanc cassé d'environ 15 mm de longueur portant l'inscription « U7 » d'un côté et aucune mention sur l'autre côté.
Plaquette thermoformable transparente en poly-chloro-tri-fluoro-éthylène (PCTFE) avec feuille d'aluminium thermoscellée-, dans une boîte contenant 7 comprimés pelliculés.
Alunbrig 90 mg comprimés pelliculés
Chaque comprimé pelliculé contient 90 mg de brigatinib.
Excipient à effet notoire
Chaque comprimé pelliculé contient 168 mg de lactose monohydraté.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Noyau du comprimé
Lactose monohydraté
Cellulose microcristalline
Carboxyméthylamidon sodique (type A)
Silice colloïdale anhydre
Stéarate de magnésium
Pelliculage
Talc
Macrogol
Alcool polyvinylique
Dioxyde de titane