
PREGABALINE ZENTIVA K.S 300 milligrammes, gélule, boîte de 56
Retiré du marché le : 23/06/2021
Dernière révision : 27/05/2021
Taux de TVA : 2.1%
Prix de vente : 19,52 €
Taux remboursement SS : 65%
Base remboursement SS : 19,52 €
Laboratoire exploitant : ZENTIVA FRANCE
Source :

Douleurs neuropathiques
Pregabaline Zentiva k.s. est indiqué dans le traitement des douleurs neuropathiques périphériques et centrales chez l'adulte.
Épilepsie
Pregabaline Zentiva k.s. est indiqué chez l'adulte en association dans le traitement des crises épileptiques partielles avec ou sans généralisation secondaire.
Trouble Anxieux Généralisé
Pregabaline Zentiva k.s. est indiqué dans le traitement du Trouble Anxieux Généralisé (TAG) chez l'adulte.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Patients
diabétiques
Conformément
aux pratiques cliniques actuelles, une adaptation du traitement hypoglycémiant
peut être nécessaire chez certains patients diabétiques ayant présenté une
augmentation de poids sous prégabaline.
Réactions
d'hypersensibilité
Des
notifications de réactions d'hypersensibilité, y compris des cas d'œdème de
Quincke, ont été rapportées après commercialisation. La survenue de symptômes
d'œdème de Quincke tels qu'un gonflement du visage, un gonflement péri-oral ou
des voies aériennes supérieures, impose l'arrêt immédiat de la prégabaline.
Etourdissements,
somnolence, perte de connaissance, confusion et altération de la fonction
mentale
Le
traitement par prégabaline a été associé à des étourdissements et de la
somnolence, qui pourraient augmenter la
survenue de blessures accidentelles (chutes) dans la population âgée. Après la
mise sur le marché, les notifications suivantes ont été rapportées : perte de
connaissance, confusion et altération de la fonction mentale. Il doit donc être
conseillé aux patients d'être prudents jusqu'à ce qu'ils soient habitués aux
effets potentiels du médicament.
Troubles de
la vision
Dans les
essais cliniques contrôlés, une proportion plus importante de patients traités
par la prégabaline que de patients sous placebo a signalé une vision trouble
qui a disparu dans la majorité des cas malgré la poursuite du traitement. Dans
les études cliniques comportant des examens ophtalmologiques, l'incidence de la
baisse de l'acuité visuelle et des modifications du champ visuel était
supérieure chez les patients du groupe prégabaline par rapport au groupe
placebo ; l'incidence des anomalies du fond d'œil était plus élevée sous
placebo (voir rubrique Propriétés pharmacodynamiques).
Au cours de l'expérience
post-commercialisation, ont également été rapportés des effets indésirables
visuels qui incluaient une perte de la vue, une vision trouble ou d'autres
modifications de l'acuité visuelle, la plupart desquels étant à caractère
transitoire. L'arrêt de la prégabaline peut entraîner la disparition de cette
symptomatologie visuelle ou son amélioration.
Insuffisance
rénale
Des cas
d'insuffisance rénale ont été rapportés et une interruption du traitement a
montré une réversibilité de cette réaction indésirable dans certains cas.
Suppression
des médicaments antiépileptiques concomitants
Il n'existe
pas de données suffisantes permettant une suppression des médicaments
antiépileptiques concomitants dans le but d'instaurer une monothérapie, lorsque
le contrôle des crises est atteint avec la prégabaline en association.
Symptômes
de sevrage
Après
interruption d'un traitement à court ou long terme par la prégabaline, des
symptômes de sevrage ont été observés chez certains patients. Les événements
suivants ont été rapportés : insomnie, céphalée, nausées, anxiété, diarrhée,
syndrome grippal, nervosité, dépression, douleurs, convulsions, hyperhidrose,
et étourdissements. Le patient doit en être informé en début de traitement.
Les
convulsions incluant les états de mal épileptiques et les états de grand mal
peuvent apparaître pendant ou peu après l'arrêt du traitement par la
prégabaline.
Concernant
l'interruption d'un traitement prolongé par la prégabaline, des données
suggèrent que l'incidence et la sévérité des symptômes de sevrage peuvent être
dose-dépendantes.
Insuffisance
cardiaque congestive
Des
notifications d'insuffisance cardiaque congestive ont été rapportées après
commercialisation chez certains patients traités par la prégabaline. Ces effets
sont observés essentiellement pendant le traitement par la prégabaline pour une
indication de douleurs neuropathiques chez les patients âgés dont la fonction
cardiovasculaire est altérée. La prégabaline doit être utilisée avec prudence
chez ces patients. Cet effet indésirable peut disparaitre à l'arrêt de la
prégabaline.
Traitement
des douleurs neuropathiques centrales dues à une lésion de la moelle épinière
Dans le
traitement des douleurs neuropathiques centrales dues à une lésion de la moelle
épinière, l'incidence des effets indésirables en général, des effets
indésirables touchant le système nerveux central et de la somnolence en
particulier, a été accrue. Ceci peut être attribué à un effet additif dû à des
médicaments concomitants (p. ex. les antispastiques) nécessaires pour ce type
d'affection. Ceci doit être pris en compte lors de la prescription de la
prégabaline pour cette affection.
Dépression
respiratoire
Des cas de
dépression respiratoire sévère ont été rapportés en lien avec l'utilisation de
la prégabaline. Les patients dont la fonction respiratoire est altérée ou
atteints d'une affection respiratoire ou d'une maladie neurologique,
d'insuffisance rénale, ou utilisant en association des médicaments dépresseurs
du système nerveux central ainsi que les personnes âgées peuvent être plus à
risque de présenter cet effet indésirable grave. Une adaptation de la posologie
peut être nécessaire pour ces patients (voir rubrique Posologie et mode
d'administration).
Idées et
comportements suicidaires
Des idées et
un comportement suicidaires ont été rapportés chez des patients traités avec
des agents antiépileptiques dans plusieurs indications. Une méta-analyse
d'essais randomisés contrôlés contre placebo de médicaments antiépileptiques a
également montré un risque légèrement accru d'idées et de comportements
suicidaires. Le mécanisme de ce risque n'est pas connu et les données
disponibles n'excluent pas la possibilité d'un risque plus élevé pour la
prégabaline.
Les patients
doivent donc être surveillés pour détecter d'éventuels signes d'idées et de
comportements suicidaires, et un traitement adapté doit être envisagé. Par
conséquent, il doit être conseillé aux patients (et aides-soignants de ces
patients) de demander un avis médical si des signes d'idées et de comportements
suicidaires apparaissent.
Ralentissement
du transit du tractus gastro-intestinal
Des
notifications d'effets indésirables liés à un ralentissement du transit du
tractus gastro-intestinal inférieur (p.ex. obstruction intestinale, iléus
paralytique, constipation) ont été rapportées après commercialisation lorsque
la prégabaline était administrée en association avec des médicaments pouvant
entraîner une constipation tels que les analgésiques opioïdes. Lorsque la
prégabaline est utilisée en association à des opioïdes, des mesures de
prévention de la constipation doivent être envisagées (en particulier chez les
femmes et les personnes âgées).
Utilisation
concomitante avec des opioïdes
La prudence
est requise lors de la prescription concomitante de prégabaline avec des
opioïdes en raison du risque de dépression du système nerveux central (SNC)
(voir rubrique Interactions avec d'autres médicaments et autres formes
d'interactions). Au cours d'une étude cas-témoins menée auprès
d'utilisateurs d'opioïdes, les patients qui prenaient de la prégabaline
conjointement avec un opioïde présentaient un risque accru de décès lié aux
opioïdes par rapport à ceux qui prenaient uniquement un opioïde (odds ratio
ajusté [ORa], 1,68 [IC à 95 %, 1,19 à 2,36]). Ce risque accru a été observé à
des doses faibles de prégabaline (≤ 300 mg, ORa 1,52 [IC 95%, 1,04 -
2,22]), et avec une tendance à l'augmentation du risque à des doses plus
élevées de prégabaline (> 300 mg, ORa 2,51 [95% IC 1,24 - 5,06]).
Abus
médicamenteux
Des cas d'abus
médicamenteux ont été rapportés. Une précaution doit être prise chez les
patients avec des antécédents de toxicomanie. Les symptômes d'abus de
prégabaline doivent être surveillés chez ces patients.
Encéphalopathie
Des cas
d'encéphalopathie ont été rapportés, principalement chez les patients
présentant des antécédents qui peuvent favoriser l'apparition d'une
encéphalopathie.
Intolérance
au lactose
Pregabaline
Zentiva k.s.contient du lactose monohydraté. Les patients présentant une
intolérance au galactose, un déficit total en lactase ou un syndrome de
malabsorption du glucose et du galactose (maladies héréditaires rares) ne
doivent pas prendre ce médicament.
Le
programme d'évaluation clinique de la prégabaline a été mené chez plus
de 8 900 patients exposés à la prégabaline, plus de 5 600 d'entre eux
l'ayant été dans le cadre d'essais en double aveugle contrôlés contre
placebo. Les effets indésirables le plus fréquemment rapportés ont été
les étourdissements et la somnolence. Ces effets étaient généralement
d'intensité légère à modérée. Dans toutes les études contrôlées, les
interruptions de traitement liées aux effets indésirables ont été de
12% pour les patients recevant la prégabaline et de 5% pour ceux
recevant le placebo. Les effets indésirables les plus fréquents ayant
entraîné l'arrêt du traitement par la prégabaline ont été les
étourdissements et la somnolence.
Le
tableau 2 ci-dessous énumère, par type et par fréquence, tous les
effets indésirables survenus à une incidence supérieure à celle du
placebo et chez plus d'un patient (très fréquent (≥ 1/10), fréquent(≥
1/100 à <1/10), peu fréquent (≥ 1/1 000 à <1/100), rare (≥ 1/10
000 à <1/1 000), très rare (<1/10 000), fréquence indéterminée
(ne peut être estimée sur la
base des données disponibles). Au sein de chaque groupe de fréquence,
les effets indésirables sont présentés par ordre de gravité
décroissante.
Les effets indésirables cités peuvent aussi être associés à la maladie sous-jacente et/ou aux médicaments concomitants.
Dans
le traitement des douleurs neuropathiques centrales dues à une lésion
de la moelle épinière, l'incidence des effets indésirables en général,
des effets indésirables touchant le SNC et de la somnolence en
particulier, a été accrue (voir rubrique Mises en garde et précautions d'emploi).
Les effets supplémentaires rapportés après commercialisation figurent dans la liste ci-dessous en italique.
Tableau 2 : Effets indésirables de la prégabaline
| Classe de systèmes d'organes | Effets indésirables |
| Infections et Infestations | |
| Peu fréquent | Nasopharyngite |
| Affections hématologiques et du système lymphatique | |
| Peu fréquent | Neutropénie |
| Affections du système immunitaire | |
| Peu fréquent | Hypersensibilité |
| Rare | Œdème de Quincke, réaction allergique |
| Troubles du métabolisme et de la nutrition | |
| Fréquent | Augmentation de l'appétit |
| Peu fréquent | Anorexie, hypoglycémie |
| Affections psychiatriques | |
| Fréquent | Humeur euphorique, confusion, irritabilité, désorientation, insomnie, diminution de la libido |
| Peu fréquent | Hallucination, attaques de panique, nervosité, agitation, dépression, humeur dépressive, agressivité, humeur changeante, dépersonnalisation, manque du mot, rêves anormaux, augmentation de la libido, anorgasmie, apathie |
| Rare | Désinhibition |
| Affections du système nerveux | |
| Très fréquent | Etourdissements, somnolence, céphalée |
| Fréquent | Ataxie, trouble de la coordination, tremblements, dysarthrie, amnésie, troubles de la mémoire, troubles de l'attention, paresthésies, hypoesthésie sédation, troubles de l'équilibre, léthargie |
| Fréquent | Syncope, stupeur, myoclonie, pertes de connaissance, hyperactivité psychomotrice, dyskinésie, vertige de position, tremblement intentionnel, nystagmus, trouble cognitif, altération de la fonction mentale, trouble du langage, hyporéflexie, hyperesthésie, sensation de brûlure, agueusie, malaise |
| Rare | Convulsions, parosmie, hypokinésie, dysgraphie |
| Affections oculaires | |
| Fréquent | Vision trouble, diplopie |
| Peu fréquent | Perte de la vision périphérique, troubles visuels, gonflement des yeux, anomalies du champ visuel, diminution de l'acuité visuelle, douleur oculaire, fatigue visuelle, photopsie, sécheresse oculaire, larmoiement, irritation des yeux |
| Classe de systèmes d'organes | Effets indésirables |
| Rare | Perte de la vue, kératite, oscillopsie, altération de la vision stéréoscopique, mydriase, strabisme, halo visuel |
| Affections de l'oreille et du labyrinthe | |
| Fréquent | Vertiges |
| Peu fréquent | Hyperacousie |
| Affections cardiaques | |
| Peu fréquent | Tachycardie, bloc auriculo-ventriculaire du premier degré, bradycardie sinusale, insuffisance cardiaque congestive |
| Rare | Allongement de l'intervalle QT, tachycardie sinusale, arythmie sinusale |
| Affections vasculaires | |
| Peu fréquent | Hypotension, hypertension, bouffées de chaleur, bouffées vasomotrices, sensation de froid aux extrémités |
| Affections respiratoires, thoraciques et médiastinales | |
| Peu fréquent | Dyspnée, epistaxis, toux, congestion nasale, rhinite, ronflement, sécheresse nasale |
| Rare | Œdème pulmonaire, sensation de constriction du pharynx |
| Fréquence indéterminée | Dépression respiratoire |
| Affections gastro-intestinales | |
| Fréquent | Vomissements, nausées, constipation, diarrhée, flatulences, distension abdominale, bouche sèche |
| Peu fréquent | Reflux gastro-œsophagien, sialorrhée, hypoesthésie orale |
| Rare | Ascite, pancréatite, gonflement de la langue, dysphagie |
| Affections hépatobiliaires | |
| Peu fréquent | Augmentation des enzymes hépatiques* |
| Rare | Ictère |
| Très rare | Insuffisance hépatique, hépatite |
| Affections de la peau et du tissu sous-cutané | |
| Peu fréquent | Eruption papuleuse, urticaire, hyperhidrose, prurit |
| Rare | Syndrome de Stevens-Johnson, sueurs froides |
| Affections musculo-squelettiques et systémiques | |
| Fréquent | Crampes musculaires, arthralgie, dorsalgie, douleur des membres, spasmes cervicaux |
| Peu fréquent | Gonflements articulaires, myalgie, contractions musculaires, douleurs cervicales, rigidité musculaire |
| Rare | Rhabdomyolyse |
| Affections du rein et des voies urinaires | |
| Peu fréquent | Incontinence urinaire, dysurie |
| Rare | Insuffisance rénale, oligurie, rétention urinaire |
| Affections des organes de reproduction et du sein | |
| Fréquent | Troubles de l'érection |
| Peu fréquent | Dysfonction sexuelle, retard de l'éjaculation, dysménorrhée, douleur mammaire |
| Rare | Aménorrhée, écoulement mammaire, hypertrophie mammaire, gynécomastie |
| Troubles généraux et anomalies au site d'administration | |
| Fréquent | Œdème périphérique, œdème, troubles de la marche, chutes, sensation d'ébriété, sensations anormales, fatigue |
| Classe de systèmes d'organes | Effets indésirables |
| Peu fréquent | Œdème généralisé, œdème de la face, oppression thoracique, douleur, hyperthermie, soif, frissons, asthénie |
| Investigations | |
| Fréquent | Prise de poids |
| Peu fréquent | Augmentation de la créatine phosphokinase sanguine, augmentation de la glycémie, numération des plaquettes diminuée, augmentation de la créatininémie, kaliémie diminuée, perte de poids |
| Rare | Numération des globules blancs diminuée |
Après
interruption d'un traitement à court ou long terme par la prégabaline,
des symptômes de sevrage ont été observés chez certains patients. Les
réactions suivantes ont été rapportées : insomnie, céphalées, nausées,
anxiété, diarrhée, syndrome grippal, convulsions, nervosité,
dépression, douleurs, hyperhidrose, et étourdissements. Le patient doit
en être informé en début de traitement.
Concernant l'interruption d'un traitement prolongé par la prégabaline, des données suggèrent que l'incidence et la sévérité des symptômes de sevrage peuvent être dose-dépendantes.
Population pédiatrique
Le
profil de sécurité d'emploi de la prégabaline observé dans cinq études
pédiatriqueschez des patients présentant des crises épileptiques
partielles avec ou sans généralisation secondaire (étude d'efficacité
et de sécurité d'emploi pendant 12semaines chez des patients âgés de 4
à 16ans, n=295; étude d'efficacité et de sécurité d'emploi pendant
14jours chez des patients âgés de 1mois à moins de 4ans, n=175; étude
de pharmacocinétique et de tolérance, n=65; et deux études de suivi de
la sécurité d'emploi en ouvert pendant 1an, n=54 et n = 431) était
similaire à celui observé dans les études menées chez les patients
adultes épileptiques. Les événements indésirables le plus fréquemment
observés au cours de l'étude de 12semaines avec le traitement par
prégabalineont été: somnolence, fièvre, infection des voies aériennes
supérieures, augmentation de l'appétit, prise de poids et
nasopharyngite. Les événements indésirables le plus fréquemment
observés au cours de l'étude de 14jours avec le traitement par
prégabalineont été: somnolence, infection des voies aériennes
supérieureset fièvre (voir rubriques4.2, 5.1 et 5.2).
Déclaration des effets indésirables suspectés
La
déclaration des effets indésirables suspectés après autorisation du
médicament est importante. Elle permet une surveillance continue du
rapport bénéfice/risque du médicament. Les professionnels de santé
déclarent tout effet indésirable suspecté via le système national de
déclaration - voir Annexe V.
SURVEILLANCE du traitement : détection d’éventuels signes d’idées et de comportements suicidaires.
CONTACTER IMMEDIATEMENT LE MEDECIN en cas de :
- gonflement du visage, des lèvres, de la langue ou de la gorge,
- rougissement de la peau et si elle commence à former des ampoules ou à peler,
- rash cutané diffus,
- vision trouble ou de perte de la vue ou d'autres modifications de la vision,
- idées autodestructrices ou suicidaires, signes de comportement suicidaire,
- diminution de la miction,
- convulsions.
- difficultés à respirer ou si la respiration semble superficielle.
EVITER la prise de boissons alcoolisées et de médicaments contenant de l'alcool pendant le traitement.
Les FEMMES EN AGE DE PROCREER doivent utiliser une contraception efficace.
NE
PAS CONDUIRE de véhicules NI UTILISER de machines NI ENTREPRENDRE
des activités potentiellement dangereuses avant d'avoir évalué
l'impact éventuel du traitement sur sa capacité à effectuer ces activités
(étourdissements, somnolence, diminution de la concentration).
Femmes en âge de procréer / Contraception chez les hommes et les femmes
Le risque encouru chez l'homme étant inconnu, les femmes en âge de procréer doivent utiliser une contraception efficace.
Grossesse
Il n'existe pas de données suffisamment pertinentes concernant l'utilisation de la prégabaline chez la femme enceinte.
Des études effectuées chez l'animal ont montré une toxicité sur la reproduction (voir rubrique Données de sécurité précliniques). Le risque potentiel en clinique n'est pas connu.
La prégabaline ne doit pas être utilisé au cours de la grossesse à moins d'une nécessité absolue (si les bénéfices pour la mère l'emportent clairement sur les risques potentiels pour le fœtus).
Allaitement
La prégabaline est excrétée dans le lait maternel (voir rubrique Propriétés pharmacocinétiques). L'effet de la prégabaline sur les nouveaux-nés/nourrissons n'est pas connu.
La décision soit d'interrompre l'allaitement soit d'interrompre le traitement avec la prégabaline doit être prise en tenant compte du bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la femme.
Fertilité
Aucune donnée clinique n'est disponible concernant les effets de la prégabaline sur la fertilité chez la femme.
Lors d'un essai clinique évaluant l'effet de la prégabaline sur la motilité des spermatozoides, les sujets hommes sains ont été exposés à une dose de 600 mg/jour. Aucun effet sur la motilité des spermatozoides n'a été observé après 3 mois de traitement.
Une étude de fertilité chez des rats femelles a montré des effets délétères sur la reproduction. Des études de fertilité chez des rats mâles ont montré des effets délétères sur la reproduction et le développement. La pertinence clinique de ces données n'est pas connue (voir rubrique Données de sécurité précliniques).
Etant donné que la prégabaline est essentiellement éliminée sous forme inchangée dans les urines, qu'elle n'est que très faiblement métabolisée chez l'homme (moins de 2% de la dose sont retrouvés dans les urines sous forme de métabolites), qu'elle n'inhibe pas le métabolisme des médicaments in vitro et qu'elle ne se lie pas aux protéines plasmatiques, celle-ci est peu susceptible d'induire ou de subir des interactions pharmacocinétiques.
Etudes in vivo et analyse pharmacocinétique de population
Par conséquent, aucune interaction pharmacocinétique cliniquement significative n'a été observée dans les études in vivo entre la prégabaline et la phénytoïne, la carbamazépine, l'acide valproïque, la lamotrigine, la gabapentine, le lorazépam, l'oxycodone ou l'éthanol. Les analyses pharmacocinétiques de population ont montré que les antidiabétiques oraux, les diurétiques, l'insuline, le phénobarbital, la tiagabine et le topiramate, n'avaient pas d'effet cliniquement significatif sur la clairance de la prégabaline.
Contraceptifs oraux, noréthistérone et/ou éthinylestradiol
L'administration concomitante de prégabaline avec les contraceptifs oraux tels que la noréthistérone et/ou l'éthinylestradiol n'influence pas les paramètres pharmacocinétiques à l'état d'équilibre de l'une ou l'autre de ces substances.
Médicaments affectant le Système Nerveux Central
La prégabaline peut potentialiser les effets de l'éthanol et du lorazépam.
Des notifications d'insuffisance respiratoire, de coma et de décès ont été rapportées après commercialisation chez des patients sous prégabaline et opioïdes et/ou autres médicaments dépresseurs du système nerveux central (SNC). L'effet de la prégabaline semble s'additionner à celui de l'oxycodone sur l'altération de la fonction cognitive et motrice globale.
Interactions et sujet âgé
Aucune étude pharmacodynamique spécifique d'interaction n'a été conduite chez les sujets âgés volontaires. Les études d'interaction n'ont été réalisées que chez l'adulte.
Posologie
La posologie varie de 150 à 600 mg par jour, en deux ou en trois prises.
Douleurs neuropathiques
Le
traitement par prégabaline peut être instauré à la dose de 150 mg par
jour administrée en deux ou en trois prises. En fonction de la réponse
et de la tolérance du patient, la dose peut être augmentée à 300 mg par
jour après un intervalle de 3 à 7 jours, et peut si nécessaire être
augmentée à la dose maximale de 600 mg par jour après un intervalle
supplémentaire de 7 jours.
Épilepsie
Le
traitement par prégabaline peut être instauré à la dose de 150 mg par
jour administrée en deux ou en trois prises. En fonction de la réponse
et de la tolérance du patient, la dose peut être augmentée à 300 mg par
jour après 1 semaine. La dose maximale de 600 mg par jour peut être
atteinte après un délai supplémentaire d'une semaine.
Trouble Anxieux Généralisé
La
posologie varie de 150 à 600 mg par jour, en deux ou en trois prises.
La nécessité de poursuivre le traitement doit être réévaluée
régulièrement.
Le traitement par prégabaline peut être instauré à la dose de 150 mg par jour. En fonction de la réponse et de la tolérance du patient, la dose peut être augmentée à 300 mg par jour après 1 semaine. Après un délai supplémentaire d'une semaine, la dose peut être augmentée à 450 mg par jour. La dose maximale de 600 mg par jour peut être atteinte après un délai supplémentaire d'une semaine.
Interruption du traitement par la prégabaline
Conformément
aux pratiques cliniques actuelles, si le traitement par la prégabaline
doit être interrompu, il est recommandé de le faire progressivement sur
une période minimale d'1 semaine quelle que soit l'indication (voir
rubriques Mises en garde et précautions d'emploi et Effets indésirables).
Insuffisants rénaux
La
prégabaline est éliminée de la circulation générale principalement par
voie rénale sous forme inchangée. La clairance de la prégabaline étant
directement proportionnelle à la clairance de la créatinine (voir
rubrique Propriétés pharmacocinétiques), chez les patients présentant une insuffisance rénale une réduction de la dose devra être établie individuellement en tenant compte de la clairance de la créatinine (CLcr), comme indiqué dans le tableau 1, calculée selon la formule suivante :
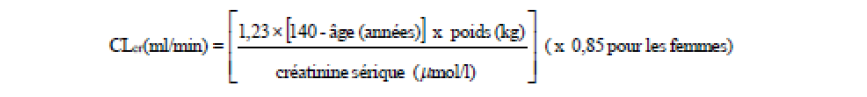
La prégabaline est éliminée efficacement du plasma par hémodialyse (50% du médicament en 4 heures). Pour les patients hémodialysés, la dose journalière de prégabaline doit être adaptée en tenant compte de la fonction rénale. En plus de la dose journalière, une dose supplémentaire doit être administrée immédiatement après chaque hémodialyse de 4 heures (voir tableau 1).
Tableau 1. Adaptation de la dose de prégabaline selon la fonction rénale
| Clairance de la créatinine (CLcr) (ml/min) | Dose journalière totale de prégabaline * | Schéma posologique | |
| | Dose Initiale (mg/jour) | Dose Maximale (mg/jour) | |
| ≥ 60 | 150 | 600 | BID ou TID |
| ≥ 30 - < 60 | 75 | 300 | BID ou TID |
| ≥ 15 - < 30 | 25 - 50 | 150 | Une fois par jour ou BID |
| < 15 | 25 | 75 | Une fois par jour |
| Dose supplémentaire après hémodialyse (mg) | |||
| | 25 | 100 | Dose unique+ |
BID = deux doses séparées
* La Dose Journalière Totale (mg/jour) doit être divisée par le nombre de prises indiqué pour obtenir le nombre de mg par prise
+ La Dose supplémentaire est une dose complémentaire administrée en une seule prise
Insuffisants hépatiques
Aucun ajustement de la dose n'est nécessaire chez les patients insuffisants hépatiques (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
La
sécurité d'emploi et l'efficacité de la prégabaline chez l'enfant de
moins de 12 ans et chez l'adolescent (12-17 ans) n'ont pas été
établies. Les données actuellement disponibles sont décrites aux
rubriques Effets indésirables, Propriétés pharmacodynamiques et Propriétés pharmacocinétiques, mais aucune recommandation sur la posologie ne peut être donnée.
Sujet âgé
En
raison d'une diminution de la fonction rénale, une réduction de la dose
de prégabaline peut être nécessaire chez les patients âgés (voir
rubrique Propriétés pharmacocinétiques).
Mode d'administration
Pregabaline
Zentiva k.s. peut être pris au moment ou en dehors des repas.
Pregabaline Zentiva k.s est administré uniquement par voie orale.
Durée de conservation :
2 ans.
Précautions particulières de conservation :
A conserver à une température ne dépassant pas 30°C.
Sans objet.
Au cours de la commercialisation, les réactions indésirables les plus fréquemment rapportées en cas de surdosage avec la prégabaline ont été : somnolence, état confusionnel, agitation et nervosité.
Des crises convulsives ont également été rapporteés.
Des cas de coma ont été rapportés dans de rares occasions.
Le traitement d'un surdosage avec la prégabaline est symptomatique et une hémodialyse peut être réalisée si nécessaire (voir rubrique Posologie et mode d'administration tableau 1).
Classe pharmacothérapeutique : Antiépileptiques, autres antiépileptiques, Code ATC : N03AX16.
La substance active, prégabaline, est un analogue [(S)-3-(aminométhyl)-5-acide méthylhexanoïque] de l'acide gamma-aminobutyrique.
Mécanisme
d'action
La prégabaline
se lie à une sous-unité auxiliaire (protéine α2-d) des canaux calciques
voltage- dépendants dans le système nerveux central.
Efficacité et sécurité clinique
Douleurs
neuropathiques
L'efficacité
de la prégabaline a été démontrée dans des essais sur la neuropathie
diabétique, la névralgie post-zostérienne et la lésion de la moelle épinière.
L'efficacité n'a pas été étudiée dans d'autres modèles de douleur
neuropathique.
La prégabaline a été étudiée au cours de 10 essais cliniques contrôlés à raison de 2 prises par jour (BID) pendant 13 semaines au maximum et de 3 prises par jour (TID) pendant 8 semaines au maximum. Dans l'ensemble, les profils de sécurité et d'efficacité ont été similaires pour les schémas posologiques BID et TID.
Dans des essais cliniques allant jusqu'à 12 semaines sur les douleurs neuropathiques périphériques et centrales, une diminution de la douleur a été observée dès la première semaine et s'est maintenue tout au long de la période de traitement.
Dans les essais cliniques contrôlés portant sur les douleurs neuropathiques périphériques, 35% des patients traités par la prégabaline et 18% des patients sous placebo ont présenté une amélioration de 50% du score de douleur. Pour les patients n'ayant pas présenté de somnolence, cette amélioration a été observée chez 33% des patients traités par la prégabaline et chez 18% des patients sous placebo. Pour les patients ayant présenté une somnolence, les taux de réponse étaient de 48% sous prégabaline et de 16% sous placebo.
Dans l'essai clinique contrôlé portant sur les douleurs neuropathiques centrales, 22 % des patients traités par la prégabaline et 7 % des patients sous placebo ont présenté une amélioration de 50 % du score de douleur.
Epilepsie
Traitement en
association
La prégabaline a été étudiée dans le cadre de 3 essais cliniques contrôlés d'une durée de 12 semaines à la posologie BID ou TID. Dans l'ensemble, les profils de sécurité et d'efficacité ont été similaires pour les schémas posologiques BID et TID.
Une diminution de la fréquence des crises a été observée dès la première semaine.
Population
pédiatrique
L'efficacité
et la sécurité d'emploi de la prégabaline n'ont pas été établies dans le
traitement en association de l'épilepsie chez les patients pédiatriques de
moins de 12 ans et chez les adolescents. Les événements indésirables chez les
patients observés lors d'une étude pharmacocinétique et de tolérance qui
incluait des patients âgés de 3 mois à 16 ans (n = 65) étaient similaires à
ceux observés chez l'adulte. Les résultats d'une étude menée versus placebo
pendant 12 semaines auprès de 295 patients pédiatriques âgés de 4 à 16 ans et
d'une étude menée versusplacebo pendant 14 jours auprès de 175 patients
pédiatriques âgés de 1 mois à moins de 4 ans portant sur l'évaluation de
l'efficacité et de la sécurité d'emploi de la prégabaline comme traitement
adjuvant des crises épileptiques partielles et de deux études de sécurité
d'emploi en ouvert pendant 1 an menées auprès de 54 et 431 patients
pédiatriques épileptiques respectivement âgés de 3 mois à 16 ans montrent que
les événements indésirables de fièvre et d'infections des voies aériennes
supérieures étaient observés plus fréquemment que dans les études chez les
patients adultes épileptiques (voir rubriques Posologie et mode d'administration,
Effets indésirables et Propriétés pharmacocinétiques).
Dans le cadre d'une étude contrôlée contre placebo pendant 12 semaines, des sujets pédiatriques (agés de 4 à 16 ans) se sont vu attribuer la prégabaline à la posologie de 2,5 mg/kg/jour (150 mg/jour au maximum), la prégabaline à la posologie de 10 mg/kg/jour (600 mg/jour au maximum), ou le placebo.
Le pourcentage de sujets ayant présenté une réduction de survenue de crises épileptiques partielles d'au moins 50 % par rapport à l'inclusion était de 40,6 % des sujets traités par la prégabaline à la posologie 13 de 10 mg/kg/jour (p = 0,0068 versus placebo), 29,1 % des sujets traités par prégabaline à la posologie de 2,5 mg/kg/jour (p = 0,2600 versus placebo) et 22,6 % de ceux recevant le placebo.
Dans le cadre d'une étude contrôlée contre placebo pendant 14jours, des sujets pédiatriques (âgés de 1mois à moins de 4ans) se sont vu attribuer la prégabaline à la posologie de 7mg/kg/jour, la prégabaline à la posologie de 14mg/kg/jour ou le placebo. Les fréquences médianes des crises épileptiques sur 24 heures étaient, respectivement, à l'inclusion et à la visite finale, de 4,7 et 3,8 pour la prégabaline 7mg/kg/jour, 5,4 et 1,4 pour la prégabaline14mg/kg/jour et 2,9 et 2,3 pour le placebo. La prégabaline14mg/kg/jour a réduit significativement la fréquence transformée logarithmiquement des crises épileptiques partielles par rapport au placebo (p=0,0223); la prégabaline7mg/kg/jour n'a pas montré d'amélioration par rapport au placebo.
Dans une étude contrôlée versus placebo de 12 semaines, 219 sujets (âgés de 5 à 65 ans, dont 66 âgés
de 5 à 16 ans) présentant des crises généralisées tonico-cloniques primaires (CTCG), ont reçu comme
traitement adjuvant de la prégabaline à la posologie de 5 mg/kg/jour (300 mg/jour au maximum), ou à
la posologie de 10 mg/kg/jour (600 mg/jour au maximum) ou le placebo. Le pourcentage de sujets
ayant présenté une réduction d'au moins 50 % du nombre de crises CTCG était respectivement de
41,3 %, 38,9 % et 41,7 % pour la prégabaline 5 mg/kg/jour, la prégabaline 10 mg/kg/jour et le
placebo.
Monothérapie
(patients nouvellement diagnostiqués)
La prégabaline
a été étudiée lors d'un essai clinique contrôlé d'une durée de 56 semaines à la
posologie BID. La prégabaline n'a pas démontré sa non-infériorité par rapport à
la lamotrigine basé sur le critère d'absence de crise pendant 6 mois. La
prégabaline et lamotrigine avaient des profils de sécurité similaires et
étaient bien tolérés.
Trouble Anxieux Généralisé
La
prégabaline a été étudiée au cours de 6 essais contrôlés d'une durée de 4 à 6
semaines, d'une étude de 8 semaines chez des sujets âgés, et d'une étude de
prévention des rechutes à long terme comportant une phase de prévention en
double aveugle d'une durée de 6 mois.
Un soulagement des symptômes du TAG, évalué par l'échelle d'anxiété de Hamilton (HAM-A) a été observé dès la première semaine.
Dans les essais cliniques contrôlés (d'une durée de 4 à 8 semaines), 52% des patients traités par la prégabaline et 38% des patients recevant un placebo ont présenté une amélioration d'au moins 50 % du score total HAM-A entre le début et la fin de l'étude.
Dans les essais cliniques contrôlés, une proportion plus importante de patients traités par la prégabaline que de patients sous placebo a signalé une vision trouble qui a disparu dans la majorité des cas malgré la poursuite du traitement. Des examens ophtalmologiques (y compris mesure de l'acuité visuelle, champ visuel standard et examen du fond d'œil avec dilatation) ont été réalisés chez plus de 3600 patients dans le cadre des essais cliniques contrôlés. Chez ces patients, 6,5 % de ceux traités par la prégabaline et 4,8 % de ceux traités par le placebo ont présenté une baisse d'acuité visuelle. Des modifications du champ visuel ont été mises en évidence chez 12,4 % des patients sous prégabaline et 11,7 % des patients recevant le placebo. Des anomalies du fond d'œil ont été observées dans 1,7 % des cas au sein du groupe prégabaline et 2,1 % dans le groupe placebo.
Les caractéristiques pharmacocinétiques à l'état d'équilibre de la prégabaline sont similaires chez les volontaires sains, chez les patients épileptiques recevant des médicaments antiépileptiques ainsi que chez les patients souffrant de douleurs chroniques.
Absorption
La prégabaline
est rapidement absorbée lorsqu'elle est administrée à jeun, les pics
plasmatiques apparaissant dans l'heure suivant l'administration d'une dose
unique ou de doses multiples. La biodisponibilité orale de la prégabaline est
estimée comme étant ≥ 90% et est indépendante de la dose. Après
administration répétée du produit, l'état d'équilibre est atteint dans un délai
de 24 à 48 heures.
Le taux
d'absorption de la prégabaline diminue lorsque le médicament est administré
avec des aliments, entraînant une diminution de la Cmax d'environ 25-30% et un
retard du tmax d'environ 2,5 heures. Toutefois, l'administration de la
prégabaline au cours du repas n'entraîne pas d'effet cliniquement significatif
sur son taux d'absorption.
Distribution
Les études
précliniques ont montré que la prégabaline traverse la barrière
hémato-encéphalique chez les souris, les rats et les singes. Il a également été
démontré que la prégabaline traverse le placenta chez les rates et est présente
dans le lait des rates allaitantes. Chez l'homme, le volume de distribution
apparent de la prégabaline après administration orale est d'environ 0,56 l/kg.
La prégabaline ne se lie pas aux protéines plasmatiques.
Métabolisme
La prégabaline
est très faiblement métabolisée chez l'homme. Après administration d'une dose
de prégabaline radio-marquée, environ 98% de la radioactivité retrouvés dans
l'urine étaient de la prégabaline sous forme inchangée. Le dérivé N-méthylé de
la prégabaline, le principal métabolite de la prégabaline retrouvé dans
l'urine, représentait 0,9% de la dose. Dans les études précliniques, aucune
racémisation de l'énantiomère S de la prégabaline en énantiomère R n'a été mise
en évidence.
Élimination
La prégabaline
est éliminée de la circulation générale principalement par voie rénale sous
forme inchangée. La demi-vie d'élimination de la prégabaline est d'environ 6,3
heures. La clairance plasmatique et la clairance rénale de la prégabaline sont
directement proportionnelles à la clairance de la créatinine (voir rubrique Propriétés
pharmacocinétiques Insuffisance rénale).
L'adaptation de la dose chez les patients ayant une fonction rénale diminuée ou traités par hémodialyse est nécessaire (voir rubrique Posologie et mode d'administration tableau 1).
Linéarité/non-linéarité
La prégabaline
présente une pharmacocinétique linéaire aux doses journalières recommandées. La
variabilité pharmacocinétique inter-individuelle observée avec la prégabaline
est faible (< 20%). La pharmacocinétique de la prégabaline administrée à
dose multiple est extrapolable à partir de celle obtenue lorsqu'elle est
administrée à dose unique. Il n'est donc pas nécessaire d'effectuer des
contrôles de routine des concentrations plasmatiques de prégabaline.
Sexe
Les études
cliniques montrent que les concentrations plasmatiques de prégabaline ne sont
pas cliniquement différentes entre les hommes et les femmes.
Insuffisance
rénale
La clairance
de la prégabaline est directement proportionnelle à la clairance de la
créatinine. De plus, la prégabaline est éliminée du plasma par hémodialyse
(après une hémodialyse de 4 heures, les concentrations plasmatiques de la
prégabaline sont réduites d'environ 50%). Etant donné que l'élimination rénale
est la voie d'élimination principale, une réduction posologique chez les
insuffisants rénaux et un complément de dose après hémodialyse s'avèrent
nécessaires (voir rubrique Posologie et mode d'administration
tableau 1).
Insuffisance
hépatique
Aucune étude
pharmacocinétique spécifique n'a été menée chez les insuffisants hépatiques.
Etant donné que la prégabaline ne subit pas de métabolisme important et qu'elle
est essentiellement excrétée sous forme inchangée dans l'urine, une
insuffisance hépatique ne devrait pas modifier significativement les
concentrations plasmatiques de prégabaline.
Population
pédiatrique
La
pharmacocinétique de la prégabaline a été évaluée chez des patients
pédiatriques épileptiques (tranches d'âge : de 1 à 23 mois, de 2 à 6 ans, de 7
à 11 ans et de 12 à 16 ans) à des niveaux de dose de 2,5, 5, 10 et 15
mg/kg/jour dans une étude pharmacocinétique et de tolérance.
Après administration orale de prégabaline chez des patients pédiatriques à jeun, le temps nécessaire pour atteindre le pic plasmatique était en général similaire dans toutes les tranches d'âge. Ce pic était atteint entre 0,5 et 2 heures après administration de la dose.
Les paramètres de Cmax et d'ASC de la prégabaline augmentaient de manière linéaire par rapport à l'augmentation de la dose dans chaque tranche d'âge. L'ASC était inférieure de 30 % chez les patients pédiatriques pesant moins de 30 kg en raison d'une plus forte clairance ajustée sur le poids corporel, de 43 %, chez ces patients par comparaison aux patients dont le poids était ≥ 30 kg.
La demi-vie terminale de la prégabaline était en moyenne de 3 à 4 heures environ chez les patients pédiatriques jusqu'à l'âge de 6 ans et de 4 à 6 heures à partir de l'âge de 7 ans.
L'analyse pharmacocinétique de population a montré que la clairance de la créatinine était une covariable significative de la clairance orale de la prégabaline, que le poids corporel était une covariable significative du volume de distribution oral apparent de la prégabaline et que ces corrélations étaient similaires chez les patients pédiatriques et adultes.
La pharmacocinétique de la prégabaline n'a pas été étudiée chez les patients de moins de 3 mois (voir rubriques Posologie et mode d'administration, Effets indésirables et Propriétés pharmacodynamiques).
Sujets âgés
La clairance
de la prégabaline tend à diminuer avec l'âge. Cette diminution de la clairance
orale de la prégabaline correspond à la diminution de la clairance de la
créatinine liée à l'âge. Une réduction de la dose de prégabaline peut s'avérer
nécessaire chez les patients qui présentent une fonction rénale diminuée en
rapport avec l'âge (voir rubrique Posologie et mode d'administration
tableau 1).
Mères
allaitantes
La
pharmacocinétique a été évaluée chez 10 femmes allaitantes recevant 150 mg de
prégabaline toutes les 12 heures (300 mg par jour), et cela au moins 12
semaines après l'accouchement. L'allaitement n'a eu que peu ou pas d'influence
sur la pharmacocinétique de la prégabaline. A l'état d'équilibre, la
prégabaline a été excrétée dans le lait maternel à des concentrations moyennes
égales à environ 76 % des concentrations plasmatiques maternelles. La quantité
ingérée par le nourrisson via le lait maternel (en supposant une
consommation de lait moyenne de 150 ml/kg/j) d'une mère recevant 300 mg/j ou la
dose maximale de 600 mg/j a été estimée respectivement à 0,31 ou 0,62 mg/kg/j.
Ces quantités correspondent à environ 7 % de la dose maternelle quotidienne
totale rapportée au poids (mg/kg).
La prégabaline peut avoir une influence mineure ou modérée sur l'aptitude à conduire des véhicules et à utiliser des machines. La prégabaline peut induire des étourdissements et une somnolence et peut donc avoir une influence sur l'aptitude à conduire ou à utiliser des machines. Il est donc conseillé aux patients de ne pas conduire, de ne pas utiliser de machines complexes ni d'entreprendre d'autres activités potentiellement dangereuses, avant d'avoir évalué l'impact éventuel de ce médicament sur leur capacité à effectuer ces activités.
Dans les études conventionnelles de pharmacologie de sécurité chez l'animal, la prégabaline a été bien tolérée à des doses cliniquement pertinentes. Dans les études de toxicité à doses répétées chez le rat et le singe, des effets sur le SNC ont été observés, parmi lesquels une hypoactivité, une hyperactivité et une ataxie. Une incidence accrue d'atrophie rétinienne communément observée chez les rats albinos âgés a été constatée après une exposition prolongée à la prégabaline ≥ 5 fois à l'exposition moyenne chez l'homme à la dose clinique maximale recommandée.
La prégabaline ne s'est pas révélée tératogène chez la souris, le rat et le lapin. Une toxicité fœtale chez le rat et le lapin est uniquement apparue lors d'expositions largement supérieures à l'exposition chez l'homme. Dans les études de toxicité pré- et postnatales, la prégabaline a induit une toxicité de la descendance chez le rat lors d'expositions > 2 fois la dose maximale recommandée chez l'homme.
Les effets indésirables observés sur la fertilité chez les rats mâles et femelles n'ont été observés qu'à des doses nettement supérieures aux doses thérapeutiques. Les effets indésirables observés sur l'appareil reproducteur mâle et sur les spermatozoides ont été réversibles et n'ont été observés qu'à des doses nettement supérieures aux doses thérapeutiques ou étaient associés à un processus dégénératif spontané de l'organe reproducteur mâle chez le rat. Ces effets sont donc considérés comme ayant peu ou pas de pertinence clinique.
La prégabaline n'est pas génotoxique comme le montrent les résultats d'une batterie de tests in vitro et in vivo.
Des études de carcinogénicité de deux ans ont été menées avec la prégabaline chez le rat et la souris. Aucune tumeur n'a été observée chez le rat lors d'expositions atteignant jusqu'à 24 fois l'exposition moyenne chez l'homme correspondant à la dose clinique maximale recommandée de 600 mg/jour.
Chez la souris, aucune augmentation de l'incidence de tumeurs n'a été observée à des expositions similaires à l'exposition moyenne chez l'homme, mais une augmentation de l'incidence des hémangiosarcomes a été observée à des expositions supérieures. Le mécanisme non génotoxique de la formation de tumeurs induite par la prégabaline chez la souris implique des modifications plaquettaires et une prolifération associée de cellules endothéliales. Ces modifications plaquettaires n'ont pas été retrouvées chez le rat ou chez l'homme, sur la base des résultats cliniques à court ou à long terme. Il n'y a aucune preuve suggérant qu'il existe un tel risque chez l'homme.
Chez le rat jeune, les données de toxicité n'étaient pas qualitativement différentes de celles observées chez le rat adulte. Les rats jeunes sont cependant plus sensibles. Aux doses thérapeutiques, des signes cliniques évidents d'hyperactivité du SNC et de bruxisme ainsi que des modifications de la croissance (suppression transitoire de la prise de poids) ont été observés. Des effets sur le cycle œstral ont été observés à des doses correspondant à 5 fois l'exposition thérapeutique chez l'homme. Une diminution de la réponse acoustique a été observée chez les rats jeunes 1 à 2 semaines après exposition à des doses 2 fois supérieures à la dose thérapeutique humaine. Neuf semaines après exposition, cet effet n'était plus observé.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
liste I
Remboursement en fonction de l'indication (JO du 15/06/2017) :
Les seules indications thérapeutiques ouvrant droit à la prise en charge ou au remboursement par l'assurance maladie sont :
- Chez l'adulte, dans le traitement des douleurs neuropathiques périphériques et centrales.
- Chez l'adulte, en association, dans le traitement des crises épileptiques partielles avec ou sans généralisation secondaire.
A partir du 24 mai 2021 :
Prescription sur ordonnance répondant aux spécifications fixées par l'arrêté du 31 mars 1999.
Prescription limitée à 6 mois de traitement.
Gélule
Gélule
composée d'une coiffe rouge et d'un corps gris clair, d'environ 21,7 mm
de long, portant l'impression « 300 » et contenant de la poudre presque
blanche.
Les gélules de Prégabaline Zentiva k.s. 300 mg sont conditionnées dans des plaquettes PVC-alu.
Les gélules de Prégabaline Zentiva k.s. 300 mg sont conditionnées dans une boite de 56 gélules.
Chaque gélule contient 300 mg de prégabaline.
Excipient à effet notoire :
Chaque gélule contient également 30 mg de lactose monohydraté.
Pour tous les excipients, voir rubrique Liste des excipients.
Contenu des
gélules
Lactose monohydraté
Amidon de maïs
prégelatinisé
Talc
Enveloppe de la gélule
Coiffe de
la gélule
Oxyde de fer
rouge (E172)
Oxyde de fer
jaune (E172)
Dioxyde de
titane (E171)
Gélatine
Corps de la gélule
Oxyde de fer
noir (E172)
Dioxyde de
titane (E171)
Gélatine
Encre d'impression
Gomme-laque
Oxyde de fer
noir (E172)
Polyéthylène
glycol