
VARUBY 90 mg, comprimé pelliculé, emballage de 2 plaquettes thermoformées de 1
Retiré du marché le : 09/09/2019
Dernière révision : 11/03/2019
Taux de TVA : 10%
Laboratoire exploitant : LILLY FRANCE
Source :

Prévention des nausées et des vomissements retardés associés à une chimiothérapie anticancéreuse hautement et modérément émétisante chez les adultes
Varuby est administré dans le cadre d'un traitement d'association (voir rubrique Posologie et mode d'administration).
Hypersensibilité à la/aux substance(s) active(s) ou à l'un des excipients mentionnés à la rubrique Composition
En association avec du millepertuis (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions)
Patients atteints d'une insuffisance hépatique sévère
Il n'existe aucune donnée sur les patients atteints d'une insuffisance hépatique sévère (voir rubrique Propriétés pharmacocinétiques). Varuby doit être utilisé avec prudence chez ces patients. Si l'utilisation ne peut pas être évitée, il conviendra de surveiller la survenue d'effets indésirables associés à Varuby chez les patients (voir rubrique Effets indésirables).
Patients atteints d'une insuffisance rénale sévère
Les données concernant les patients atteints d'une insuffisance rénale sévère sont limitées (voir rubrique Propriétés pharmacocinétiques). Varuby doit être utilisé avec précaution chez ces patients. Si l'utilisation ne peut pas être évitée, il conviendra de surveiller la survenue d'effets indésirables associés à Varuby chez les patients (voir rubrique Effets indésirables).
Interactions
Varuby est déconseillé chez les patients nécessitant une administration chronique d'inducteurs enzymatiques puissants (ex. : la rifampicine, la carbamazépine, le phénobarbital, l'enzalutamide, la phénytoïne) ou modérés (ex. : l'éfavirenz, la rifabutine) (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
L'efficacité et la sécurité du rolapitant utilisé en association avec un autre antagoniste des récepteurs NK1 (ex. : aprépitant et une combinaison de nétupitant et de chlorhydrate de palonosétron) n'ont pas été établies et une telle utilisation est par conséquent déconseillée (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Lactose
Varuby contient du lactose. Les patients souffrant de troubles héréditaires rares, comme une intolérance au galactose, un déficit en lactase de Lapp ou un syndrome de malabsorption du glucose- galactose, ne doivent pas prendre ce médicament.
Résumé du profil de sécurité
Plus de 4 375 sujets ont été traités par Varuby ou un comparateur dans le cadre d'études cliniques de phases 1, 2 et 3. Au total, 2 798 sujets ont reçu du rolapitant par voie orale, quelle que soit la dose, incluant 1 567 patients au cours des études portant sur les nausées et les vomissements chimio-induits (NVCI).
Les effets indésirables les plus fréquents ont été la fatigue (1,9 %) et les céphalées (1,5 %). Le profil de sécurité lors des phases d'extension à cycles multiples des études portant sur des chimiothérapies hautement et moyennement émétisantes, jusqu'à 6 cycles de chimiothérapie, a été similaire à celui observé au Cycle 1.
Liste des effets indésirables sous forme de tableau
Les effets indésirables suivants ont été observés lors d'une analyse combinée des études sur les chimiothérapies hautement émétisantes (CHE) et les chimiothérapies modérément émétisantes (CME).
La fréquence est indiquée comme suit : « très fréquent » (≥ 1/10), « fréquent » (≥ 1/100, < 1/10), « peu fréquent » (≥ 1/1 000, < 1/100), « rare » (≥ 1/10 000, < 1/1 000) ou « très rare » (< 1/10 000), fréquence indéterminée : ne peut être estimée sur la base des données disponibles.
|
Effets indésirables par classe de systèmes d'organes | |||
|
Classe de systèmes d'organes |
Fréquent |
Peu fréquent |
Rare |
|
Infections et infestations |
|
Mycose orale |
Candidose Candidose orale |
|
Affections hématologiques et du système lymphatique |
|
Neutropénie |
INR augmenté (International Normalised Ratio) Leucopénie Neutrophiles diminués Thrombopénie |
|
Affections du système immunitaire |
|
|
Hypersensibilité |
|
Troubles du métabolisme et de la nutrition |
|
Appétit diminué |
Déshydratation Hypomagnésémie |
|
Affections psychiatriques |
|
Insomnie |
Anxiété Bruxisme |
|
Affections du système nerveux |
Céphalée |
Sensation vertigineuse Perturbation de l'attention Dysgueusie Somnolence |
Troubles de l'équilibre Perturbation des mouvements Syncope |
|
Affections de l'oreille et du labyrinthe |
|
|
Hypoacousie Acouphènes |
|
Affections oculaires |
|
|
Vision trouble |
|
Affections cardiaques |
|
|
Fréquence cardiaque augmentée |
|
Affections gastro- intestinales |
Constipation |
Diarrhée Dyspepsie Nausées Distension abdominale Douleurs abdominales Stomatite |
Gêne abdominale Changement d'habitude de transit Bouche sèche Reflux gastro-œsophagien Efforts de vomissement |
|
Affections vasculaires |
|
|
Hypertension |
|
Affections respiratoires, thoraciques et médiastinales |
|
Hoquet |
Dyspnée |
|
Effets indésirables par classe de systèmes d'organes | |||
|
Classe de systèmes d'organes |
Fréquent |
Peu fréquent |
Rare |
|
Affections de la peau et du tissu sous-cutané |
|
|
Alopécie Angiœdème Dermatite acnéiforme Peau sèche |
|
Affections musculo- squelettiques et systémiques |
|
Myalgie |
Arthralgie Dorsalgie Faiblesse musculaire Rhabdomyolyse |
|
Troubles généraux et anomalies au site d'administration |
Fatigue |
Asthénie |
Troubles de la démarche |
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
PREVENIR IMMEDIATEMENT LE MEDECIN OU L'INFIRMIER/ERE en cas de réaction allergique :
- Difficultés respiratoires soudaines.
- Gonflement des lèvres ou de la langue ou une altération du goût.
- Gonflement de la peau ou des tissus ou une éruption cutanée soudaine ou de la fièvre.
- Accélération du rythme cardiaque.
NE PAS CONSOMMER de produits à base de plantes contenant du millepertuis (Hypericum perforatum).
PRUDENCE en cas de conduite de véhicules ou d'utilisation de machines (sensations vertigineuses, fatigue).
Grossesse
Il n'existe aucune donnée disponible sur l'utilisation de rolapitant chez la femme enceinte. Les études chez l'animal n'ont révélé aucun effet tératogène ou embryo-fœtal. Lors d'une étude sur le développement pré- et post-natal, à une dose équivalente à environ la moitié de la dose maximale recommandée chez l'homme, une baisse de mémoire des ratons femelles lors d'un test de labyrinthe ainsi qu'une diminution du poids corporel des ratons ont été observées (voir rubrique Données de sécurité précliniques). Varuby ne doit pas être utilisé pendant la grossesse, sauf en cas de nécessité absolue.
Allaitement
Il n'existe aucune donnée sur la présence du rolapitant dans le lait maternel humain. Le rolapitant administré par voie orale chez des rates en lactation était présent dans le lait. L'allaitement est déconseillé pendant tout traitement avec Varuby.
Fertilité
Le rolapitant n'a pas affecté la fertilité ou la capacité reproductive générale des rats mâles. Des diminutions du nombre de corps jaunes et de sites d'implantation ont été observées lors d'une étude sur la fertilité des rates et le développement embryonnaire précoce (voir rubrique Données de sécurité précliniques).
Effets de Varuby sur la pharmacocinétique d'autres substances actives
Substrats du CYP2D6
Le rolapitant est un inhibiteur modéré du CYP2D6. Une concentration plasmatique accrue de substrats du CYP2D6 peut entraîner des effets indésirables potentiels. Une multiplication par 3 de l'exposition du dextrométhorphane, un substrat du CYP2D6, a été observée 7 jours après la prise d'une dose unique de rolapitant par voie orale et pourrait persister plus longtemps.
Par conséquent, il convient de faire preuve de prudence lorsque le rolapitant est associé à un médicament métabolisé par le CYP2D6, notamment ceux ayant une marge thérapeutique étroite (ex. : la propafénone, le tamoxifène, le métoprolol utilisé dans le traitement de l'insuffisance cardiaque, la thioridazine, le pimozide).
Substrats UGT1A1 et UGT2B7 (par exemple l'irinotécan et la morphine, respectivement)
Le rolapitant inhibait modestement l'UGT1A1 et l'UGT2B7 in vitro. Par conséquent, les interactions potentielles associées à l'inhibition de ces enzymes UGT au niveau intestinal ne peuvent être exclues.
Substrats de la BCRP
Le rolapitant est un inhibiteur de la protéine de résistance au cancer du sein (« breast cancer resistance protein », BCRP). Des concentrations plasmatiques accrues de substrats de la BCRP (ex. : le méthotrexate, l'irinotécan, le topotécan, la mitoxantrone, la rosuvastatine, la sulfasalazine, la doxorubicine, la bendamustine) pourraient entraîner des effets indésirables potentiels. L'administration concomitante d'une dose unique de 180 mg de rolapitant avec de la sulfasalazine, un substrat de la BCRP, a entraîné une multiplication par 2 environ de la Cmax et de l'ASC de la sulfasalazine. Si l'association ne peut être évitée, une surveillance clinique et biologique des effets indésirables liés au médicament concomitant doit être mise en place. La rosuvastatine doit être utilisée à la dose efficace la plus faible.
Substrats de la P-gp
Le rolapitant est un inhibiteur de la P-glycoprotéine (P-gp). Une augmentation de 70 % de la Cmax et de 30 % de l'ASC de la digoxine, un substrat de la P-gp, a été observée lorsque celle-ci a été administrée avec une dose unique de 180 mg de rolapitant. Par conséquent, il conviendra d'effectuer une surveillance clinique des effets indésirables chez le patient et, si possible, une surveillance biologique, en cas d'utilisation concomitante de rolapitant et de digoxine ou d'autres substrats de la P-gp (ex. : le dabigatran ou la colchicine), en particulier chez les patients atteints d'insuffisance rénale.
Substrats d'OATP1B1 et d'OATP1B3
Les études in vitro suggèrent que le rolapitant ne devrait pas inhiber l'OATP1B1 à des concentrations cliniquement pertinentes et le rolapitant n'est pas un inhibiteur de l'OATPB1B3 aux concentrations testées, jusqu'à 20 µM.
Substrats de l'OCT1
In vitro, le rolapitant n'est pas un inhibiteur de l'OCT1 aux concentrations testées, jusqu'à 20 µM.
Substrats du CYP3A4
In vivo, le rolapitant ne devrait pas avoir d'effet inhibiteur ou inducteur sur le CYP3A4. Une dose unique de 180 mg de rolapitant n'a eu aucun effet significatif sur la pharmacocinétique du midazolam par rapport à 3 mg de midazolam administré seul par voie orale le Jour 1, le Jour 8 et le Jour 11.
Ondansétron
Le rolapitant n'a eu aucun effet significatif sur la pharmacocinétique de l'ondansétron administré par voie intraveineuse lors d'une administration concomitante avec une dose unique de 180 mg de rolapitant le même jour.
Dexaméthasone
Le rolapitant n'a eu aucun effet significatif sur la pharmacocinétique de la dexaméthasone lorsque de la dexaméthasone a été administrée les Jours 1 à 3 par voie orale, après l'administration concomitante d'une dose unique de 180 mg de rolapitant le Jour 1.
Substrats d'autres CYP
Aucune interaction cliniquement significative n'est attendue avec les médicaments suivants lors d'une administration avec une dose unique de 180 mg de rolapitant le Jour 1 et sans rolapitant le Jour 8 : le répaglinide 0,25 mg (un substrat du CYP2C8), l'éfavirenz 600 mg (un substrat du CYP2B6), le tolbutamide 500 mg (un substrat du CYP2C9) ou l'oméprazole 40 mg (un substrat du CYP2C19).
Le rolapitant n'a montré aucun effet sur la pharmacocinétique de la caféine (un substrat du CYP1A2) après administration d'une dose orale de 200 mg de caféine avec une dose unique de 180 mg de rolapitant le Jour 1 et sans rolapitant le Jour 8 et le Jour 15.
Effets d'autres médicaments sur la pharmacocinétique de Varuby
Inducteurs enzymatiques
L'administration concomitante de rifampicine, un inducteur enzymatique puissant, a réduit de manière significative l'exposition systémique au rolapitant et à son métabolite actif. Lorsque 600 mg de rifampicine ont été administrés une fois par jour pendant 7 jours avant et 7 jours après l'administration d'une dose unique de 180 mg de rolapitant, l'ASC moyenne a été réduite de 87 % et celle de son métabolite actif de 89 % par rapport à l'administration de rolapitant seul. Varuby est déconseillé chez les patients nécessitant une administration chronique de puissants inducteurs enzymatiques (ex. : la rifampicine, la carbamazépine, l'enzalutamide, la phénytoïne) (voir rubrique Mises en garde et précautions d'emploi).
L'effet des inducteurs modérés (ex. : l'éfavirenz, la rifabutine) n'est pas établi ; par conséquent, l'utilisation de rolapitant chez les patients auxquels un inducteur modéré est déjà administré est déconseillée (voir rubrique Mises en garde et précautions d'emploi).
Du fait de son effet inducteur puissant, le millepertuis est contre-indiqué avec le rolapitant (voir rubrique Contre-indications).
Inhibiteurs du CYP3A4
Aucun effet cliniquement significatif n'a été observé sur la pharmacocinétique du rolapitant lorsque du kétoconazole, un puissant inhibiteur du CYP3A4, a été administré avec du rolapitant. Une administration concomitante de 400 mg de kétoconazole une fois par jour pendant 21 jours, à la suite d'une dose unique de 90 mg de rolapitant, n'a pas eu d'effet significatif sur la Cmax du rolapitant, alors que l'ASC a augmenté de 21 %. Cela ne devrait pas avoir de conséquences cliniques pertinentes.
Autres interactions
L'efficacité et la sécurité du rolapitant utilisé en concomitance avec un autre antagoniste des récepteurs NK1 (ex. : aprépitant et une combinaison de nétupitant et de chlorure de palonosétron) n'ont pas été établies et une telle utilisation est par conséquent déconseillée (voir rubrique Mises en garde et précautions d'emploi).
Posologie
Adultes
Varuby doit être administré dans le cadre d'un schéma thérapeutique comportant de la dexaméthasone et un antagoniste des récepteurs 5-HT3.
180 mg (deux comprimés) doivent être administrés par voie orale dans les 2 heures précédant le début de chaque cycle de chimiothérapie, mais à des intervalles d'au moins 2 semaines entre chaque prise.
Il n'y a pas d'interaction médicamenteuse entre le rolapitant et la dexaméthasone ; aucun ajustement posologique n'est par conséquent requis pour la dexaméthasone.
Les schémas thérapeutiques suivants sont recommandés pour la prévention des nausées et des vomissements associés à un traitement anticancéreux émétisant :
Schéma de chimiothérapie hautement émétisante
| Jour 1 | Jour 2 | Jour 3 | Jour 4 | |
| Varuby | 180 mg par voie orale ; dans les 2 heures précédant la chimiothérapie | Aucun | ||
| Dexaméthasone | 20 mg par voie orale ; 30 min avant la chimiothérapie | 8 mg par voie orale, deux fois par jour | 8 mg par voie orale, deux fois par jour | 8 mg par voie orale, deux fois par jour |
| Antagoniste des récepteurs 5-HT3 | Dose standard de l'antagoniste des récepteurs 5-HT3. Voir le résumé des caractéristiques du produit pour connaître la posologie appropriée pour l'antagoniste des récepteurs 5-HT3 administré en association. | Aucun | ||
Schéma de chimiothérapie modérément émétisante
| Jour 1 | Jour 2 | Jour 3 | Jour 4 | |
| Varuby | 180 mg par voie orale ; dans les 2 heures précédant la chimiothérapie | Aucun | ||
| Dexaméthasone | 20 mg par voie orale ; 30 min avant la chimiothérapie | Aucun | ||
| Antagoniste des récepteurs 5-HT3 | Dose standard de l'antagoniste des récepteurs 5-HT3. Voir le résumé des caractéristiques du produit pour connaître la posologie appropriée pour l'antagoniste des récepteurs 5-HT3 administré en association. | Voir le résumé des caractéristiques du produit pour connaître la posologie appropriée pour l'antagoniste des récepteurs 5-HT3 administré en association. | ||
Populations particulières
Personnes âgées (≥ 65 ans)
Aucun ajustement posologique n'est nécessaire pour les personnes âgées. Les données disponibles chez les patients âgés de 75 ans sont limitées. Par conséquent, Varuby doit être utilisé avec précaution chez ces patients. (voir rubrique Propriétés pharmacocinétiques).
Insuffisance rénale
Aucun ajustement posologique n'est nécessaire pour les patients atteints d'une insuffisance rénale légère ou modérée. Les données concernant les patients avec une atteinte grave de la fonction rénale sont limitées et aucune donnée n'existe chez les patients atteints d'une maladie rénale terminale sous hémodialyse Varuby doit être utilisé avec prudence chez ces patients (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
Aucun ajustement posologique n'est nécessaire pour les patients atteints d'une insuffisance hépatique légère ou modérée. Il n'existe aucune donnée sur les patients atteints d'une insuffisance hépatique sévère. Varuby doit être utilisé avec prudence chez ces patients (voir rubriques Mises en garde et précautions d'emploi et Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l'efficacité du rolapitant chez les enfants et les adolescents de moins de 18 ans n'ont pas encore été établies. Aucune donnée n'est disponible.
Mode d'administration
Les comprimés doivent être avalés entiers, avec de l'eau et peuvent être pris avec ou sans nourriture.
Durée de conservation :
4 ans
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
Des doses de rolapitant allant jusqu'à 720 mg ont été utilisées lors d'études cliniques, sans signaux de sécurité. En cas de surdosage, l'administration du médicament doit être interrompue et un traitement de support adapté et une surveillance du patient doivent être mis en place. Du fait de l'activité antiémétisante du rolapitant, l'activité émétisante d'autres médicaments pourrait ne pas être efficace. Aucune étude de dialyse n'a été effectuée.
Classe pharmacothérapeutique : Antiémétiques et antinauséeux, autres antiémétiques, code ATC : A04AD14
Mécanisme d'action
Le rolapitant est un antagoniste sélectif des récepteurs de la substance P humaine/neurokinine 1 (NK1).
Efficacité et sécurité cliniques
Chimiothérapie hautement émétisante (CHE) à base de cisplatine
Étude 1 et Étude 2 (CHE)
Dans le cadre de deux études cliniques multicentriques de phase III, randomisées, en double aveugle, contrôlées, en groupes parallèles, (Étude 1 et Étude 2), le schéma thérapeutique du rolapitant (180 mg de rolapitant, 10 µg/kg de granisétron par voie intraveineuse et 20 mg de dexaméthasone par voie orale) a été comparé au traitement du bras témoin (placebo, 10 µg/kg de granisétron par voie intraveineuse et 20 mg de dexaméthasone par voie orale) le Jour 1, chez des patients recevant une chimiothérapie comportant du cisplatine ≥ 60 mg/m2. Les patients ont reçu 8 mg de dexaméthasone par voie orale, deux fois par jour les Jours 2 à 4. Les médicaments de l'étude ont été administrés avant la chimiothérapie le Jour 1 aux intervalles suivants : 1 à 2 heures avant pour le rolapitant, et 30 minutes avant pour le granisétron et la dexaméthasone.
Au total, 1 087 patients ont été randomisés pour recevoir soit le traitement par rolapitant (N = 544), soit le traitement du bras témoin (N = 543) au cours de l'Étude 1 et de l'Étude 2 ; 1 070 patients ont été inclus dans l'évaluation de l'efficacité - 37 % étaient des femmes et 63 % étaient des hommes. Parmi ces 1 070 patients, 26 % avaient plus de 65 ans et 3 % avaient plus de 75 ans.
Le critère d'évaluation primaire pour les deux études était la réponse complète (définie comme l'absence d'épisodes émétiques et le non-recours à un médicament de secours) dans la phase retardée (> 24 à 120 heures) de nausées et de vomissements induits par la chimiothérapie. Les critères d'évaluation secondaires pré-spécifiés suivants ont également été évalués : la réponse complète durant la phase aiguë (0 à 24 heures) et durant la phase globale (0 à 120 heures) ; l'absence de vomissements dans chaque phase de NVCI ; l'absence de nausées significatives dans chaque phase de NVCI et le délai jusqu'à la survenue du 1er vomissement ou le recours à un médicament de secours.
Les résultats ont été évalués pour chaque étude et pour les deux études combinées. Les résultats individuels des Études 1 et 2 et un résumé des principaux résultats pour l'analyse combinée sont présentés dans le Tableau 1 ci-après.
| Tableau 1 : Proportion de patients recevant une chimiothérapie à base de cisplatine et répondant au traitement en fonction des groupes et des phases de traitement (Études 1 et 2 - Résultats individuels sur la CHE) | |||||||||
| Critères d'évaluation de l'efficacitéa | Étude 1 sur la CHE | Étude 2 sur la CHE | Études 1 et 2 combinées | ||||||
| Taux (en %) de patients traités par rolapitant (N = 264) | Taux (en %) de patients traités dans le groupe témoin (N = 262) | pb | Taux (en %) de patients traités par rolapitant (N = 271) | Taux (en %) de patients traités dans le groupe témoin (N = 273) | pb | Taux (en %) de patients traités par rolapitant (N = 535) | Taux (en %) de patients traités dans le groupe témoin (N = 535) | pc | |
| Réponse complète | |||||||||
| Phase retardée | 72,7 | 58,4 | < 0,001 | 70,1 | 61,9 | 0,043 | 71,4 | 60,2 | < 0,001 |
| Phase aiguë | 83,7 | 73,7 | 0,005 | 83,4 | 79,5 | n. s. | 83,6 | 76,6 | 0,004 |
| Phase globale | 70,1 | 56,5 | 0,001 | 67,5 | 60,4 | n. s. | 68,8 | 58,5 | < 0,001 |
| Aucun vomissement | |||||||||
| Phase aiguë | 86,4 | 76,0 | 0,002 | 85,6 | 81,7 | n. s. | 86,0 | 78,9 | 0,002 |
| Phase retardée | 78,0 | 61,8 | < 0,001 | 73,1 | 65,2 | 0,046* | 75,5 | 63,6 | < 0,001 |
| Phase globale | 75,4 | 59,2 | < 0,001 | 70,8 | 64,1 | n. s. | 73,1 | 61,7 | < 0,001 |
| Aucune nausée significative | |||||||||
| Phase aiguë | 86,4 | 79,4 | 0,035 | 90,0 | 85,7 | n. s. | 88,2 | 82,6 | 0,009 |
| Phase retardée | 73,5 | 64,9 | 0,034 | 74,5 | 68,9 | n. s. | 74,0 | 66,9 | 0,011 |
| Phase globale | 71,6 | 63,0 | 0,037 | 72,7 | 67,8 | n. s. | 72,1 | 65,4 | 0,017 |
| a Le critère d'évaluation primaire était la réponse complète au cours de la phase tardive. Phase tardive : > 24 à 120 heures après le traitement à base de cisplatine ; phase aiguë : > 0 à 24 heures après le traitement à base de cisplatine ; phase globale : 0 à 120 heures après le traitement à base de cisplatine b Les valeurs p non ajustées sont obtenues à partir du test de Cochran-Mantel-Haenszel, stratifié en fonction du sexe. c Les valeurs p non ajustées sont obtenues à partir du test de Cochran-Mantel-Haenszel, stratifié en fonction du sexe et de l'étude. n. s. = non significatif (p > 0,05) *Non significatif après application d'un ajustement pour multiplicité préalablement défini. | |||||||||
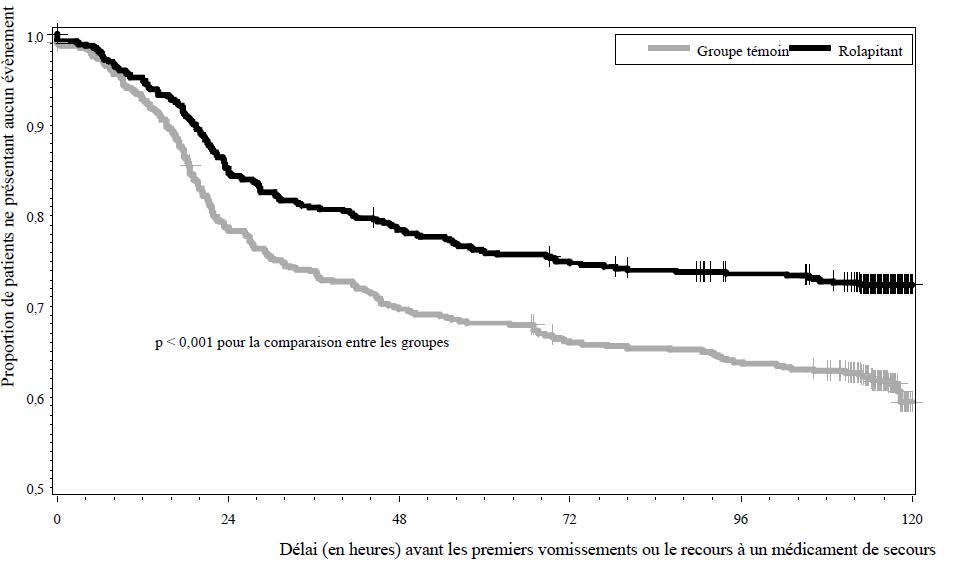
Le délai estimé jusqu'à la survenue du premier vomissement dans l'analyse combinée est illustré par la courbe de Kaplan-Meier en figure 1.
Figure 1 : Courbe de Kaplan-Meier relative à la proportion de patients sans vomissements ni recours à un médicament de secours (Étude 1 et Étude 2 combinées sur la CHE)
Chimiothérapie modérément émétisante et chimiothérapie associant une anthracycline et le cyclophosphamide
Étude 3 (CME)
Dans l'Étude 3, une étude clinique multicentrique, randomisées, en double aveugle, contrôlées, en groupes parallèles, portant sur la chimiothérapie modérément émétisante, le schéma thérapeutique du rolapitant (180 mg de rolapitant, 2 mg de granisétron par voie orale et 20 mg de dexaméthasone par voie orale) a été comparé au traitement du bras témoin (placebo, 2 mg de granisétron par voie orale et 20 mg de dexaméthasone par voie orale) le Jour 1, chez des patients recevant une chimiothérapie modérément émétisante, 53 % d'entre eux recevant une association d'anthracycline et de cyclophosphamide (AC). Les Jours 2 à 3, les patients ont reçu 2 mg de granisétron par voie orale, une fois par jour. Les médicaments de l'étude ont été administrés avant la chimiothérapie le Jour 1 aux intervalles suivants : 1 à 2 heures avant pour le rolapitant, et 30 minutes avant pour le granisétron et la dexaméthasone. Au moment de la conception de l'étude, les schémas de chimiothérapie contenant une association AC étaient considérés comme modérément émétisants. Les récentes recommandations therapeutiques ont réévalué ces schémas et les ont classifié comme hautement émétisants. Le pourcentage de patients ayant reçu du carboplatine lors du Cycle 1 était de 30 %.
Au total, 1 369 patients ont été randomisés pour recevoir soit le traitement par rolapitant (N = 684), soit le traitement du bras témoin (N = 685) ; 1 332 patients ont été inclus dans l'évaluation de l'efficacité ; 80 % étaient des femmes et 20 % étaient des hommes. Parmi ces 1 332 patients, 28 % avaient plus de 65 ans, 6 % plus de 75 ans et 629 d'entre eux ont reçu une chimiothérapie non-AC.
Le critère d'évaluation primaire était la réponse complète (définie comme l'absence d'épisodes émétiques et le non-recours à un médicament de secours) dans la phase retardée (> 24 à 120 heures) de nausées et de vomissements induits par la chimiothérapie. Les critères d'évaluation secondaires pré- spécifiés suivants ont également été évalués :la réponse complète au cours de la phase aiguë (0 à 24 heures) et de la phase globale (0 à 120 heures) ; l'absence de vomissements dans chaque phase de NVCI ; l'absence de nausées significatives dans chaque phase de NVCI et le délai jusqu'à la survenue du 1er vomissement ou avant le recours à un médicament de secours.
Un résumé des résultats de l'étude sur la CME (Étude 3) est présenté dans le Tableau 2 ci-après. Un résumé des résultats des sous-populations non-AC et AC est présenté dans le Tableau 3.
| Tableau 2 : Proportion de patients recevant une chimiothérapie modérément émétisante et répondant au traitement en fonction des groupes et des phases de traitement | |||
| Critères d'évaluation de l'efficacitéa | Étude 3 (CME) | ||
| Taux (en %) de patients traités par rolapitant (N = 666) | Taux (en %) de patients traités par rolapitant (N = 666) | pb | |
| Réponse complète Phase retardée Phase aiguë Phase globale | 71,3 83,5 68,6 | 61,6 80,3 57,8 | < 0,001 n. s. < 0,001* |
| Aucun vomissement | | | |
| Phase aiguë | 87,8 | 84,5 | n. s. |
| Phase retardée | 80,5 | 69,8 | < 0,001* |
| Phase globale | 78,7 | 65,3 | < 0,001* |
| Aucune nausée significative (score EVA maximal < 25 sur une échelle allant de 0 à 100) | | | |
| Phase aiguë | 82,1 | 84,7 | n. s. |
| Phase retardée | 72,7 | 69,4 | n. s. |
| Phase globale | 70,6 | 66,5 | n. s. |
| a Le critère d'évaluation primaire était la réponse complète au cours de la phase tardive. Phase aiguë : 0 à 24 heures après le schéma AC ou non-AC ; Phase tardive : > 24 à 120 heures après le schéma AC ou non-AC ; Phase globale : 0 à 120 heures après le schéma AC ou non-AC b Les valeurs p non ajustées sont obtenues à partir du test de Cochran-Mantel-Haenszel, stratifié en fonction du sexe. n. s. = non significatif (p > 0,05) *Non significatif après application d'un ajustement pour multiplicité préalablement défini. | |||
| Tableau 3 : Proportion de patients recevant une chimiothérapie AC ou non-AC et obtenant une réponse complète | |||
| Réponse complète | Taux (en %) | Taux (en %) | pc |
| Non-AC Phase retardée Phase aiguë Phase globale | N = 322 76,1 90,7 74,8 | N = 307 63,8 84,4 61,2 | < 0,001 0,016 < 0,001 |
| AC Phase retardée Phase aiguë Phase globale | N = 344 66,9 76,7 62,8 | N = 359 59,6 76,9 54,9 | 0,047 n. s. 0,033 |
| a Les valeurs p non ajustées sont obtenues à partir du test Cochran-Mantel-Haenszel. n. s. = non significatif (p > 0,05) | |||
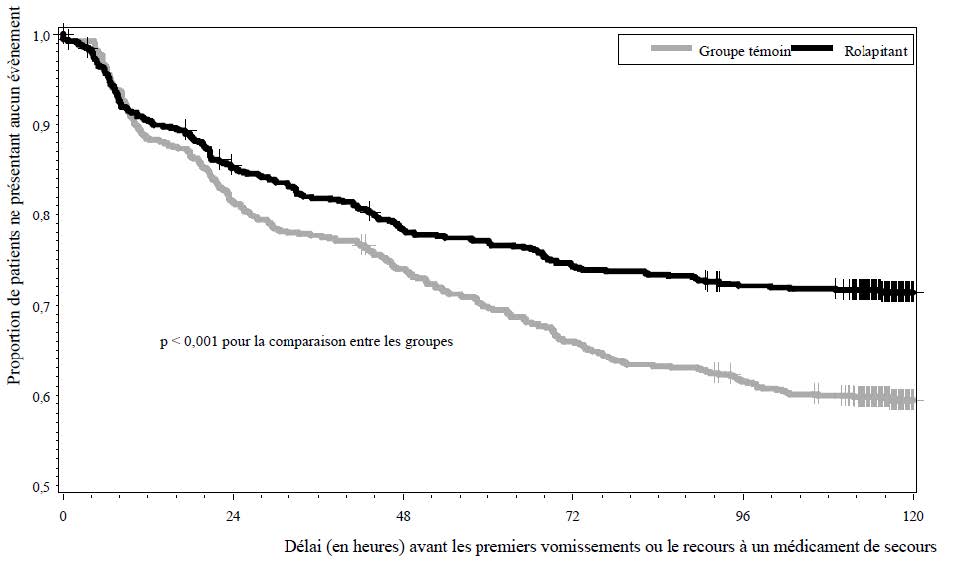
Le délai estimé jusqu'à la survenue du 1er vomissement ou jusqu'au recours à un médicament de secours chez les patients recevant un schéma de CME est illustré par la courbe de Kaplan-Meier de la figure 2.
Figure 2 : Courbe de Kaplan-Meier relative à la proportion de patients sans vomissements ni recours à un médicament de secours (Étude 3-- sur la CME)
L'impact des nausées et des vomissements sur la vie quotidienne des patients a été évalué à l'aide du questionnaire Functional Living Index-Emesis (FLIE). La proportion de patients chez lesquels les nausées et les vomissements n'ont eu aucun impact sur la vie quotidienne était plus élevée dans le groupe traité par Varuby que dans le groupe témoin (CME : 73,2 % contre 67,4 % ; p = 0,027).
Phase d'extension à cycles multiples : dans chaque étude, les patients avaient la possibilité de poursuivre dans une phase d'extension sur plusieurs cycles jusqu'à 5 cycles supplémentaires de chimiothérapie, en recevant le même schéma de traitement que celui attribué au Cycle 1. Aux Jours 6 à 8 après le début de la chimiothérapie, il a été demandé aux patients s'ils avaient eu un épisode de vomissement, de haut-le-cœur ou de nausée ayant eu un impact sur leur vie quotidienne habituelle.
L'activité antiémétique du rolapitant a été maintenue pendant les cycles répétés pour les patients poursuivant dans chacun des cycles multiples.
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats des études réalisées avec le rolapitant dans tous les sous-groupes de la population pédiatrique en prévention des nausées et des vomissements aigus et tardifs associés au cycle initial et subséquents de chimiothérapie hautement émétisante à base de cisplatine et de chimiothérapie modérément émétisante (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Le rolapitant présente une pharmacocinétique linéaire, avec l'augmentation des expositions qui est proportionnelle à la dose. Le rolapitant est éliminé lentement, avec une demi-vie terminale moyenne d'environ 7 jours. Le rolapitant est principalement éliminé par voie hépatobiliaire et est éliminé de façon mineure par les reins. Le rolapitant est métabolisé par le CYP3A4 pour former un métabolite actif majeur, le M19. Les études in vitro suggèrent que le rolapitant n'est pas un inhibiteur du CYP2E1.
Absorption
Après administration d'une dose unique de 180 mg de rolapitant chez des sujets sains et à jeun, le rolapitant a été mesurable dans le plasma entre 30 minutes et la concentration plasmatique maximale (Cmax) du rolapitant, qui a été atteinte en 4 heures environ. La valeur Cmax moyenne était de 968 ng/ml (% du CV : 28 %). Après l'administration répétée par voie orale de doses journalières de 9 à 45 mg de rolapitant, l'accumulation de rolapitant a été multipliée par 5 environ.
Les expositions systémiques (Cmax et ASC) du rolapitant ont augmenté proportionnellement à la dose lorsque la dose de rolapitant a été augmentée de 4,5 mg à 180 mg. Avec une multiplication par 4 de la dose par rapport à la dose clinique recommandée de 180 mg, la Cmax et l'ASC du rolapitant ont été multipliées par 3,1 et 3,7, respectivement.
La biodisponibilité absolue du rolapitant est d'environ 100 %, indiquant un effet minime de premier passage.
L'administration concomitante d'un repas riche en graisses n'a pas affecté de manière significative la pharmacocinétique du rolapitant après l'administration de 180 mg de rolapitant.
Distribution
Le rolapitant était fortement lié aux protéines dans le plasma humain (99,8 %). Le volume de distribution apparent (Vd/F) était de 460 L chez les sujets sains, indiquant une distribution tissulaire étendue du rolapitant. Dans une analyse pharmacocinétique de population du rolapitant, le Vd/F était de 387 L chez les patients atteints de cancer.
Biotransformation
Le
rolapitant est métabolisé par le CYP3A4 pour former un métabolite actif
majeur, le M19 (rolapitant hydroxylé par la pyrrolidine en C4).- Lors
d'une étude d'équilibre de masse, le métabolite M19 était le principal
métabolite en circulation. La formation de M19 a été retardée de
manière significative avec un tmax moyen
de 120 heures (intervalle : 24-168 heures) et une demi-vie moyenne du
M19 de 158 heures. Le rapport d'exposition du M19 au rolapitant était
d'environ 50 % dans le plasma.
Élimination
Après prise unique par voie orale (de 4,5 à 180 mg) de rolapitant, la demi-vie terminale moyenne (t½) du rolapitant a varié entre 169 et 183 heures (7 jours environ) et a été indépendante de la dose. Dans une analyse pharmacocinétique de population, la clairance totale apparente (Cl/F) du rolapitant était de 0,96 L/heure chez les patients atteints de cancer.
Le rolapitant est éliminé principalement par voie hépatobiliaire. Après l'administration par voie orale d'une dose unique de 180 mg de rolapitant marqué au [14C], en moyenne, 14,2 % (intervalle : 9 à 20 %) de la dose a été retrouvée dans les urines, et 73 % (intervalle : 52 à 89 %) de la dose a été retrouvée dans les selles sur 6 semaines. Dans des échantillons regroupés recueillis sur 2 semaines, 8,3 % de la dose a été retrouvée dans les urines, principalement sous forme de métabolites, et 37,8 % de la dose a été retrouvée dans les selles, principalement sous forme inchangée de rolapitant. Le rolapitant ou le M19, sous forme inchangée, n'ont pas été retrouvés dans les échantillons d'urine regroupés. Les enzymes métabolisant le médicament (et les transporteurs du médicament) autres que le CYP3A4 impliqué dans l'élimination hépatobiliaire du rolapitant restent à élucider.
Pharmacocinétique chez les populations particulières
Âge, sexe et origine ethnique
Les analyses de pharmacocinétique de populations ont indiqué que l'âge, le sexe et l'origine ethnique n'avaient pas d'impact significatif sur la pharmacocinétique de Varuby. Les données concernant les patients âgés de 75 ans et plus sont limitées.
Insuffisance hépatique
Après administration d'une dose unique de 180 mg de rolapitant chez des patients atteints d'une insuffisance hépatique légère (classe A de Child-Pugh), la pharmacocinétique du rolapitant était comparable à celle de sujets sains. Chez des patients atteints d'une insuffisance hépatique modérée (classe B de Child-Pugh), la Cmax moyenne était inférieure à 25 %, alors que l'ASC moyenne du rolapitant était similaire à celle des sujets sains. Le tmax médian pour le M19 a été retardé à 204 heures chez les patients atteints d'une insuffisance hépatique légère ou modérée contre 168 heures chez les sujets sains. La pharmacocinétique de Varuby n'a pas été étudiée chez les patients atteints d'une insuffisance hépatique sévère (classe C de Child-Pugh).
Insuffisance rénale
Dans des analyses de pharmacocinétique de population, la clairance de la créatinine (ClCr) à l'inclusion n'a pas montré d'effet significatif sur la pharmacocinétique du rolapitant chez les patients atteints de cancer et souffrant d'une insuffisance rénale légère (ClCr : 60 à 90 mL/min) ou modérée (ClCr : 30 à 60 mL/min), par rapport aux patients atteints d'un cancer avec une fonction rénale normale. Les informations sont insuffisantes en ce qui concerne l'effet d'une insuffisance rénale sévère. La pharmacocinétique de Varuby n'a pas été étudiée chez les patients atteints d'une maladie rénale terminale sous hémodialyse.
Relation effet / dose
Occupation des récepteurs NK1
Une étude de tomographie par émission de positons (TEP) chez l'homme et portant sur le rolapitant a démontré que le rolapitant traverse la barrière hémato-encéphalique et occupe les récepteurs NK1 du cerveau. Une augmentation dose-dépendante du taux moyen d‘occupation des récepteurs NK1 a été observée avec des posologies allant de 4,5 mg à 180 mg de rolapitant. À des concentrations plasmatiques de rolapitant > 15 ng/mL et 348 ng/mL, les taux d'occupation des récepteurs NK1 dans les régions corticales ont été d'environ > 50 % et 90 %, respectivement. À la dose de 180 mg de rolapitant, le taux moyen d'occupation des récepteurs NK1 dans les régions corticales était supérieur à 90 % pendant au moins 120 heures.
Varuby a un impact mineur sur l'aptitude à conduire des véhicules et à utiliser des machines. Des sensations vertigineuses et de la fatigue peuvent survenir après l'administration de rolapitant (voir rubrique Effets indésirables).
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, de génotoxicité, du potentiel carcinogène et de toxicité sur la reproduction, ne révèlent pas de danger particulier pour l'homme.
Le mécanisme expliquant la différence significative des demi-vies observées entre le rat et le singe (6-8 h) et l'homme (7 jours) n'est pas élucidé.
Le rolapitant a été testé chez les rongeurs lors d'études de toxicité à doses orales répétées allant jusqu'à 26 semaines et le foie, la thyroïde, les reins, l'épididyme et l'utérus ont été identifiés comme organes cibles. Lors d'une étude de trois mois réalisée chez le rat, des convulsions cloniques ont été observées chez un seul animal à 125 mg/kg/jour (environ 6 fois la dose humaine recommandée exprimée en surface corporelle). Lors d'une étude d'un mois réalisée chez le singe, des convulsions ont été observées à 60 mg/kg/jour (environ 5,8 fois la dose humaine recommandée exprimée en surface corporelle). La pertinence des convulsions chez l'homme est inconnue.
Lors d'une étude sur la fertilité et le développement embryonnaire précoce chez les rates, l'administration d'une dose orale équivalent à 9 mg/kg par jour de chlorhydrate de rolapitant base libre (environ 0,5 fois la dose humaine recommandée exprimée en surface corporelle) a entraîné une baisse transitoire du gain de poids des mères et une augmentation de l'incidence de pertes pré- et post- implantation. À une dose équivalent à 4,5 mg/kg par jour de base libre (environ 0,2 fois la dose humaine recommandée exprimée en surface corporelle), des diminutions du nombre de corps jaune et de sites d'implantation ont été observées.
Lors d'une étude de développement pré- et postnatal chez le rat, la toxicité maternelle a été observée sur la base de la mortalité / morbidité, la diminution du poids corporel et de la consommation de nourriture, la perte de la totalité de la portée, la mise bas prolongée, la durée de gestation réduite et l'augmentation de la perte du nombre de sites d'implantation, à une dose équivalent à 22,5 mg/kg par jour de rolapitant base libre (environ 1,2 fois la dose humaine recommandée exprimée en surface corporelle). Les effets sur la progéniture à cette dose ont inclus une diminution de la survie postnatale, du poids corporel et du gain pondéral ; ils pourraient être liés à la toxicité maternelle observée. À une dose maternelle équivalent à 9 mg/kg par jour de rolapitant base libre (environ 0,5 fois la dose humaine recommandée exprimée en surface corporelle), il a été observé une baisse de la mémoire lors d'un test de labyrinthe chez des jeunes rats femelles et une diminution du poids corporel des jeunes rats.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Les comprimés sont bleus et imprimés « T0101 » sur une face et « 100 » sur l'autre.
Double plaquette thermoformée en polychlorure de vinyle/polychlorotrifluoroéthylène/feuille d'aluminium.
Emballage contenant deux comprimés.