
KEYTRUDA 25 mg-mL, solution à diluer pour perfusion, boîte de 1 flacon de 4 ml
Dernière révision : 16/03/2023
Taux de TVA : 2.1%
Laboratoire exploitant : MSD FRANCE
Source :

Mélanome
KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes et des adolescents âgés de 12 ans et plus atteints d'un mélanome avancé (non résécable ou métastatique).
KEYTRUDA est indiqué en monothérapie dans le traitement adjuvant des patients adultes et des adolescents âgés de 12 ans et plus atteints d'un mélanome de stade IIB, IIC ou III, ayant eu une résection complète (voir rubrique Propriétés pharmacodynamiques).
Cancer bronchique non à petites cellules (CBNPC)
KEYTRUDA est indiqué en monothérapie dans le traitement de première ligne des patients adultes atteints d'un cancer bronchique non à petites cellules métastatique dont les tumeurs expriment PD-L1 avec un score de proportion tumorale (TPS) ≥50 %, sans mutations tumorales d'EGFR ou d'ALK.
KEYTRUDA, en association à une chimiothérapie pemetrexed et sel de platine, est indiqué dans le traitement de première ligne des patients adultes atteints de cancer bronchique non à petites cellules métastatique non-épidermoïde dont les tumeurs ne présentent pas de mutations d'EGFR ou d'ALK.
KEYTRUDA, en association au carboplatine et au paclitaxel ou au nab-paclitaxel, est indiqué dans le traitement de première ligne des patients adultes atteints de cancer bronchique non à petites cellules métastatique épidermoïde.
KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes atteints de cancer bronchique non à petites cellules localement avancé ou métastatique dont les tumeurs expriment PD-L1 avecun TPS ≥ 1 %, et ayant reçu au moins une chimiothérapie antérieure. Les patients présentant des mutations tumorales d'EGFR ou d'ALK doivent également avoir reçu une thérapie ciblée avant de recevoir KEYTRUDA.
Lymphome de Hodgkin classique (LHc)
KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes et pédiatriques âgés de 3 ans et plus atteints d'un lymphome de Hodgkin classique en rechute ou réfractaire après échec d'une greffe de cellules souches (GCS) autologue ou après au moins deux lignes de traitement antérieures lorsque la GCS autologue n'est pas une option de traitement.
Carcinome urothélial
KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes atteints d'un carcinome urothélial localement avancé ou métastatique ayant reçu une chimiothérapie antérieure à base de sels de platine (voir rubrique Propriétés pharmacodynamiques).
KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes atteints d'un carcinome urothélial localement avancé ou métastatique inéligibles à une chimiothérapie à base de cisplatine et dont les tumeurs expriment PD-L1 avec un score positif combiné (CPS) ≥ 10 (voir rubrique Propriétés pharmacodynamiques).
Carcinome épidermoïde de la tête et du cou (CETEC)
KEYTRUDA est indiqué en monothérapie ou en association à une
chimiothérapie à base de sels de platine et de 5-fluorouracile (5-FU) dans le traitement de première ligne des patients adultes atteints d'un carcinome épidermoïde de la tête et du cou métastatique ou récidivant non résécable dont les tumeurs expriment PD-L1 avec un CPS ≥ 1 (voir rubrique Propriétés pharmacodynamiques).
KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes atteints d'un carcinome épidermoïde de la tête et du cou récidivant ou métastatique dont les tumeurs expriment PD-L1 avec un TPS ≥ 50 % et en progression pendant ou après une chimiothérapie à base de sels de platine (voir rubrique Propriétés pharmacodynamiques).
Carcinome à cellules rénales (CCR)
KEYTRUDA, en association à l'axitinib, est indiqué dans le traitement de première ligne des patients adultes atteints d'un carcinome à cellules rénales avancé (voir rubrique Propriétés pharmacodynamiques).
KEYTRUDA, en association au lenvatinib, est indiqué dans le traitement de première ligne des patients adultes atteints d'un carcinome à cellules rénales avancé (voir rubrique Propriétés pharmacodynamiques).
KEYTRUDA est indiqué en monothérapie dans le traitement adjuvant des patients adultes atteints d'un carcinome à cellules rénales à risque accru de récidive post néphrectomie, ou après une néphrectomie et une résection des lésions métastatiques (pour les critères de sélection, veuillez voir la rubrique Propriétés pharmacodynamiques).
Cancers avec instabilité microsatellitaire élevée (MSI-H) ou déficience du système de réparation des mésappariements de l'ADN (dMMR)
Cancer colorectal
KEYTRUDA est indiqué en monothérapie chez des patients adultes atteints d'un cancer colorectal MSI-H ou dMMR aux stades suivants :
- traitement de première ligne d'un cancer colorectal métastatique ;
- traitement d'un cancer colorectal non résécable ou métastatique après
traitement antérieur à base de fluoropyrimidine en association.
Cancers non-colorectaux
KEYTRUDA est indiqué en monothérapie dans le traitement des patients adultes atteints de tumeurs MSI-H ou dMMR suivantes :
- cancer de l'endomètre avancé ou récidivant, dont la maladie progresse
pendant ou après un traitement antérieur à base de sels de platine reçu
quel que soit le stade et qui ne sont pas éligibles à une chirurgie
curative ou à une radiothérapie ;
- cancer gastrique, de l'intestin grêle ou des voies biliaires non
résécable ou métastatique, dont la maladie progresse pendant ou après
au moins un traitement antérieur.
Cancer de l'œsophage
KEYTRUDA, en association à une chimiothérapie à base de sels de platine
et de fluoropyrimidine, est indiqué dans le traitement de première
ligne des patients adultes atteints d'un cancer de l'œsophage ou d'un adénocarcinome de la jonction gastro-œsophagienne HER-2 négatif, localement avancés non résécables ou métastatiques, dont les tumeurs expriment PD-L1 avec un CPS ≥ 10 ( voir section Propriétés pharmacodynamiques).
Cancer du sein triple négatif (CSTN)
KEYTRUDA, en association à une chimiothérapie comme traitement
néoadjuvant, puis poursuivi après la chirurgie en monothérapie comme
traitement adjuvant, est indiqué dans le traitement des patients
adultes atteints d'un cancer du sein triple négatif localement avancé
ou de stade précoce à haut risque de récidive (voir rubrique Propriétés pharmacodynamiques).
KEYTRUDA, en association à une chimiothérapie, est indiqué dans le traitement des patients adultes atteints d'un cancer du sein triple négatif localement récurrent non résécable ou métastatique, dont les tumeurs expriment PD-L1 avec un CPS ≥ 10 et qui n'ont pas reçu de chimiothérapie antérieure pour la maladie métastatique (voir section Propriétés pharmacodynamiques).
Cancer de l'endomètre (CE)
KEYTRUDA, en association au lenvatinib, est indiqué dans le traitement des patientes adultes atteintes d'un cancer de l'endomètre avancé ou récidivant, dont la maladie progresse pendant ou après un traitement antérieur
à base de sels de platine reçu quel que soit le stade et qui ne sont
pas éligibles à une chirurgie curative ou à une radiothérapie.
Cancer du col de l'utérus
KEYTRUDA, en association à une chimiothérapie avec ou sans bevacizumab, est indiqué dans le traitement des patientes adultes atteintes d'un cancer du col de l'utérus persistant, récidivant ou métastatique, dont les tumeurs expriment PD-L1 avec un CPS ≥ 1.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Evaluation du statut PD-L1
Lors de l'évaluation du statut PD-L1 de la tumeur, il est important de choisir une méthodologie robuste et validée pour minimiser les faux-négatifs et les faux-positifs.
Effets indésirables d'origine immunologique
Des effets indésirables d'origine immunologique, y compris des cas sévères et d'issue fatale, ont été rapportés chez des patients recevant pembrolizumab. La plupart des effets indésirables d'origine immunologique apparus au cours du traitement par pembrolizumab ont été réversibles et pris en charge par une interruption de pembrolizumab, l'administration de corticostéroïdes et/ou des soins de support. Des effets indésirables d'origine immunologique ont également été rapportés après la dernière administration de pembrolizumab. Des effets indésirables d'origine immunologique affectant plus d'un système d'organes peuvent survenir simultanément.
En cas de suspicion d'effets indésirables d'origine immunologique, il convient de faire une évaluation appropriée pour confirmer l'étiologie ou éliminer les autres causes. En fonction de la sévérité de l'effet indésirable, pembrolizumab doit être suspendu et des corticostéroïdes doivent être administrés. En cas d'amélioration au Grade ≤ 1, une diminution progressive de la corticothérapie doit être initiée et poursuivie sur une période d'au moins 1 mois. Sur la base de données limitées issues des études cliniques chez les patients dont les effets indésirables d'origine immunologique n'ont pu être contrôlés par des corticostéroïdes, l'administration d'autres immunosuppresseurs systémiques peut être envisagée.
Si l'effet indésirable s'améliore jusqu'au Grade ≤ 1 et si la dose de corticostéroïdes est réduite à ≤ 10 mg de prednisone ou équivalent par jour, pembrolizumab peut être repris dans les 12 semaines suivant la dernière administration de KEYTRUDA.
En cas d'effet indésirable d'origine immunologique de Grade 3 récurrent et pour toute toxicité due à un effet indésirable d'origine immunologique de Grade 4, pembrolizumab doit être arrêté définitivement, à l'exception des endocrinopathies contrôlées par traitement hormonal substitutif(voir rubriques Posologie et mode d'administration et Effets indésirables).
Pneumopathie inflammatoire d'origine immunologique
Des cas de pneumopathie inflammatoire ont été rapportés chez des patients recevant pembrolizumab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour rechercher les signes et symptômes de pneumopathie inflammatoire. Une pneumopathie inflammatoire suspectée doit être confirmée par une évaluationradiologique et les autres causes doivent être éliminées. En cas d'évènements de Grade ≥ 2, des corticostéroïdes doivent être administrés (dose initiale de 1-2 mg/kg par jour de prednisone ou équivalent, puis diminution progressive). En cas de pneumopathie inflammatoire de Grade 2, pembrolizumab doit être suspendu. En cas de pneumopathie inflammatoire de Grade 3, de Grade 4 ou récurrente de Grade 2, pembrolizumab doit être arrêté définitivement (voir rubrique Posologie et mode d'administration).
Colite d'origine immunologique
Des cas de colite ont été rapportés chez des patients recevant pembrolizumab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour rechercher les signes et symptômes de colite et les autres causes doivent être éliminées. En cas d'évènements de Grade ≥ 2, des corticostéroïdes doivent être administrés (dose initiale de 1-2 mg/kg par jour de prednisone ou équivalent, puis diminution progressive). En cas de colite de Grade 2 ou de Grade 3, pembrolizumab doit être suspendu. En cas de colite de Grade 4 ou colite récurrente de Grade 3, pembrolizumab doit être arrêté définitivement (voir rubrique Posologie et mode d'administration). Le risque potentiel de perforation gastro-intestinale doit être pris en considération.
Hépatite d'origine immunologique
Des cas d'hépatite ont été rapportés chez des patients recevant pembrolizumab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour rechercher des modifications de la fonction hépatique (à l'initiation du traitement, régulièrement pendant le traitement et en fonction de l'évaluation clinique) et les symptômes d'une hépatite ; les autres causes doivent être éliminées. Des corticostéroïdes doivent être administrés (dose initiale de 0,5-1 mg/kg de prednisone ou équivalent par jour pour les évènements de Grade 2, et dose initiale de 1-2 mg/kg par jour pour les évènements de Grade ≥ 3, puis diminution progressive) et, selon la sévérité de l'augmentation des enzymes hépatiques, pembrolizumab doit être suspendu ou arrêté définitivement (voir rubrique Posologie et mode d'administration).
Néphrite d'origine immunologique
Des cas de néphrite ont été rapportés chez des patients recevant pembrolizumab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour rechercher des modifications de la fonction rénale et les autres causes de dysfonctionnement rénal doivent être éliminées. En cas d'ef ets indésirables de Grade ≥ 2, des corticostéroïdes doivent être administrés (dose initiale de 1-2 mg/kg par jour de prednisone ou équivalent, puis diminution progressive). En fonction de la sévérité de l'augmentation de la créatinine : en cas de néphrite de Grade 2, pembrolizumab doit être suspendu ; en cas de néphrite de Grade 3 ou de Grade 4, pembrolizumab doit être arrêté définitivement (voir rubrique Posologie et mode d'administration).
Endocrinopathies d'origine immunologique
Des endocrinopathies sévères incluant insuffisance surrénalienne, hypophysite, diabète de type 1, acidocétose diabétique, hypothyroïdie et hyperthyroïdie ont été observées au cours du traitement par pembrolizumab.
En cas d'endocrinopathies d'origine immunologique, un traitement hormonal substitutifà long terme peut être nécessaire.
Une insuffisance surrénalienne (primaire et secondaire) a été rapportée chez des patients recevant pembrolizumab. Des cas d'hypophysite ont aussi été rapportés chez des patients recevant pembrolizumab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour rechercher les signes et symptômes d'une insuffisance surrénalienne et d'une hypophysite (y compris un hypopituitarisme) et les autres causes doivent être éliminées. Des corticostéroïdes doivent être administrés pour traiter l'insuffisance surrénalienne, et d'autres traitements hormonaux substitutifs doivent être administrés selon les indications cliniques. En cas d'insuffisance surrénalienne ou d'hypophysite de Grade 2, pembrolizumab doit être suspendu jusqu'au contrôle de l'évènement par traitement hormonal substitutif. Pembrolizumab doit être suspendu ou arrêté en cas d'insuffisance surrénalienne ou d'hypophysite symptomatique de Grades 3 ou 4. La poursuite de pembrolizumab peut être envisagée après diminution progressive de la corticothérapie, si nécessaire (voir rubrique Posologie et mode d'administration). La fonction hypophysaire et les taux hormonaux doivent être surveillés pour s'assurer que le traitement hormonal substitutif est approprié.
Des cas de diabète de type 1, y compris d'acidocétose diabétique, ont été rapportés chez des patients recevant pembrolizumab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour rechercher une hyperglycémie ou d'autres signes et symptômes du diabète. De l'insuline doit être administrée pour le diabète de type 1. En cas de diabète de type 1 associé à une acidocétose ou à une hyperglycémie de Grade ≥ 3, pembrolizumab doit être suspendu jusqu'à obtention du contrôle métabolique (voir rubrique Posologie et mode d'administration).
Des
troubles thyroïdiens, y compris hypothyroïdie, hyperthyroïdie et
thyroïdite, ont été rapportés chez des patients recevant pembrolizumab
et peuvent survenir à tout moment pendant le traitement. L'hypothyroïdie est plus fréquemment rapportée chez les patients atteints de CETEC préalablement traités par une radiothérapie. Les patients doivent être surveillés pour rechercher des modifications de la fonction thyroïdienne
(à l'initiation du traitement, régulièrement pendant le traitement et
en fonction de l'évaluation clinique) et des signes et symptômes
cliniques de troubles thyroïdiens. L'hypothyroïdie peut être prise encharge avec un traitement hormonal substitutif sans interruption de traitement et sans corticostéroïdes.
L'hyperthyroïdie peut être prise en charge de façon symptomatique. En
cas d'hyperthyroïdie de Grade ≥ 3, pembrolizumab doit être suspendu
jusqu'à amélioration à Grade ≤ 1. La fonction thyroïdienne et les taux hormonaux doivent être surveillés pour s'assurer que le traitement hormonal substitutifest approprié.
Pour les patients atteints d'endocrinopathies de Grade 3 ou de Grade 4 qui s'améliorent jusqu'au Grade 2 ou inférieur, et sont contrôlées par un traitement hormonal substitutif, si indiqué, la poursuite de pembrolizumab peut être envisagée après diminution progressive de la corticothérapie, si nécessaire. Sinon, le traitement doit être arrêté (voir rubriques Posologie et mode d'administration et Effets indésirables).
Effets indésirables cutanés d'origine immunologique
Des réactions cutanées sévères d'origine immunologique ont été rapportées chez des patients recevant pembrolizumab (voir rubrique Effets indésirables). Les patients doivent être surveillés pour des réactions cutanées sévères suspectées et les autres causes doivent être éliminées. En fonction de la sévérité de l'effet indésirable, pembrolizumab doit être suspendu en cas de réactions cutanées de Grade 3 jusqu'à amélioration au Grade ≤ 1 ou arrêté définitivement en cas de réactions cutanées de Grade 4, et des corticoïdes doivent être administrés (voir rubrique Posologie et mode d'administration).
Des cas de syndrome de Stevens-Johnson (SSJ) et de nécrolyse épidermique toxique (NET) ont été rapportés chez des patients recevant pembrolizumab (voir rubrique Effets indésirables). En cas de suspicion de SSJ ou de NET, pembrolizumab doit être suspendu et le patient doit être dirigé vers une unité spécialisée pour évaluation et traitement. Si le SSJ ou la NET sont confirmés, pembrolizumab doit être arrêté définitivement (voirrubrique Posologie et mode d'administration).
La prudence s'impose lorsque l'utilisation de pembrolizumab est envisagée chez un patient ayant déjà présenté un effet indésirable cutané sévère ou engageant le pronostic vital lors d'un précédent traitement avec d'autres agents anticancéreux stimulant le système immunitaire.
Autres efets indésirables d'origine immunologique
Les ef ets indésirables supplémentaires suivants, d'origine immunologique et cliniquement significatifs ont été rapportés dans les études cliniques ou depuis la commercialisation : uvéite, arthrite, myosite, myocardite, pancréatite, syndrome de Guillain-Barré, syndrome myasthénique, anémie hémolytique, sarcoïdose, encéphalite, myélite, vascularite, cholangite sclérosante, gastrite, cystite non infectieuse et hypoparathyroïdie (voir rubriques Posologie et mode d'administration et Effets indésirables).
Selon la sévérité et le type des effets indésirables, pembrolizumab doit être suspendu en cas d'événements de Grade 2 ou de Grade 3 et des corticoïdes administrés.
Pembrolizumab peut être repris dans les 12 semaines suivant la dernière administration de KEYTRUDA si l'effet indésirable est amélioré jusqu'au Grade ≤ 1 et si la dose de corticostéroïdes a été réduite à ≤ 10 mg de prednisone ou équivalent par jour.
En cas d'ef et indésirable d'origine immunologique de Grade 3 récurrent et pour tout effet indésirable d'origine immunologique de Grade 4, pembrolizumab doit être arrêté définitivement.
En cas de myocardite, d'encéphalite ou de syndrome de Guillain-Barré de Grades 3 ou 4, pembrolizumab doit être arrêté définitivement (voir rubriques Posologie et mode d'administration et Effets indésirables).
Effets indésirables liés à une greffe
Rejet de grefe d'organe solide
Un rejet de greffe d'organe solide a été signalé après la mise sur le marché chez des patients traités par inhibiteurs du PD-1. Le traitement par pembrolizumab peut augmenter le risque de rejet chez les bénéficiaires d'une greffe d'organe solide. Il convient de prendre en considération le rapport entre les bénéfices du traitement par pembrolizumab et le risque de rejet d'organe chez ces patients.
Complications d'une greffe de cellules souches hématopoïétiques (GCSH) allogénique
GCSH allogénique après traitement par pembrolizumab
Des cas de réaction du greffe on contre l'hôte (GVH) et de maladie veino-occlusive (MVO) hépatique ont été observés chez des patients atteints d'un LHc ayant reçu une GCSH allogénique après une exposition antérieure au pembrolizumab. Jusqu'à la mise à disposition de nouvelles données, une évaluation attentive des
bénéfices potentiels d'une GCSH et d'une possible augmentation du
risque de complications liées à la greffe doit être effectuée au cas
par cas (voir rubrique Effets indésirables).
GCSH allogénique avant traitement par pembrolizumab
Chez des patients avec un antécédent de GCSH allogénique, une GVH
aiguë, y compris une GVH mortelle, a été rapportée après traitement par
pembrolizumab. Les patients ayant présenté une GVH après leur greffe
peuvent avoir un risque plus élevé de développer une GVH après
traitement par pembrolizumab. Evaluer lebénéfice du traitement par pembrolizumab par rapport au risque d'une possible GVH chez les patients avec un antécédent de GCSH allogénique.
Réactions liées à la perfusion
Des cas de réactions sévères liées à la perfusion, incluant des cas d'hypersensibilité et des cas d'anaphylaxie, ont été rapportés chez des patients recevant pembrolizumab (voir rubrique Effets indésirables). En cas de réactions à la perfusion de Grades 3 ou 4, la perfusion doit être arrêtée et pembrolizumab doit être arrêté définitivement (voir rubrique Posologie et mode d'administration). Les patients ayant présenté des réactions à la perfusion de Grades 1 ou 2 peuvent continuer à recevoir pembrolizumab sous surveillance étroite ; une prémédication par antipyrétique et antihistaminique peut être envisagée.
Utilisation du pembrolizumab en association à une chimiothérapie
Pembrolizumab en association à une chimiothérapie doit être utilisé avec prudence chez les patients âgés de ≥ 75 ans, en considérant attentivement et au cas par cas le rapport bénéfice/risque potentiel (voir section Propriétés pharmacodynamiques).
Précautions spécifiques à la maladie
Utilisation du pembrolizumab dans le carcinome urothélial chez les patients ayant reçu une chimiothérapie antérieure à base de sels de platine
Les médecins doivent prendre en considération l'efficacité retardée du pembrolizumab avant d'initier le traitement chez les patients ayant des facteurs pronostiques défavorables et/ou une maladie agressive. Dans le carcinome urothélial, un nombre plus élevé de décès dans les 2 premiers mois a été observé avec le pembrolizumab en comparaison à la chimiothérapie (voir rubrique Propriétés pharmacodynamiques). Les facteurs associés à ces décès précoces étaient une progression rapide de la maladie lors d'un traitement antérieur à base de sels de platine et la présence de métastases hépatiques.
Utilisation du pembrolizumab dans le carcinome urothélial chez les patients étant considérés comme inéligibles à une chimiothérapie à base de cisplatine et dont les tumeurs expriment PD-L1 avec un CPS ≥ 10 Les caractéristiques à l'inclusion et les facteurs pronostiques de la pathologie dans la population de l'étude
KEYNOTE-052 incluaient une proportion de patients éligibles à une association à base de carboplatine pour lesquels le bénéfice a été évalué dans une étude comparative (KEYNOTE-361). Dans l'étude KEYNOTE- 361, un nombre plus élevé de décès dans les 6 mois d'initiation du traitement suivi d'un bénéfice de survie à long terme ont été observés avec le pembrolizumab en monothérapie par rapport à la chimiothérapie (voir rubrique Propriétés pharmacodynamiques). Aucun facteur spécifique associé aux décès précoces n'a pu être identifié. Les médecins doivent prendre en considération l'effet différé du pembrolizumab avant d'initier le traitement chez les patients atteints d'un carcinome urothélial considérés comme éligibles à une association de chimiothérapie àbase de carboplatine. L'étude KEYNOTE-052 incluait également des patients éligibles à une mono-chimiothérapie, chez lesquels aucune donnée randomisée n'est disponible. De plus, il n'existe pas de données disponibles sur la sécurité et l'efficacité chez les patients plus fragiles (par exemple, statut de performance ECOG de 3) considérés comme non éligibles à une chimiothérapie. En l'absence de ces données, pembrolizumab doit être utilisé avec prudence dans cette population, après évaluation attentive du rapport bénéfice/risque potentiel au cas par cas.
Utilisation du pembrolizumab pour le traitement de première ligne des patients atteints de CBNPC
En général, la fréquence des effets indésirables du traitement par pembrolizumab en association est plus élevée que pour pembrolizumab en monothérapie ou pour la chimiothérapie seule, reflétant la contribution de chacun de ces composants (voir rubriques Posologie et mode d'administration et Effets indésirables). Il n'existe pas de comparaison directe de pembrolizumab administré en association à une chimiothérapie et de pembrolizumab en monothérapie.
Les médecins doivent considérer le rapport bénéfice/risque des options de traitement disponibles (pembrolizumab en monothérapie ou pembrolizumab en association à la chimiothérapie) avant d'instaurer le traitement chez les patients atteints de CBNPC non préalablement traités dont les tumeurs expriment PD-L1.
Dans l'étude KEYNOTE-042, il a été observé un nombre plus élevé de décès dans les 4 mois d'initiation du traitement suivi d'un bénéfice de survie à long terme avec le pembrolizumab en monothérapie par rapport à la chimiothérapie (voir rubrique Propriétés pharmacodynamiques).
Utilisation du pembrolizumab dans le traitement de première ligne des patients atteints d'un CETEC
En général, la fréquence des effets indésirables pour le traitement par pembrolizumab en association est plus élevée que pour pembrolizumab en monothérapie ou pour la chimiothérapie seule, reflétant la contribution de chacun de ces composants (voir rubrique Effets indésirables).
Les médecins doivent considérer le rapport bénéfice/risque des options de traitement disponibles (pembrolizumab en monothérapie ou pembrolizumab en association à la chimiothérapie) avant d'instaurer le traitement chez les patients atteints d'un CETEC dont les tumeurs expriment PD-L1 (voir rubrique Propriétés pharmacodynamiques).
Utilisation du pembrolizumab pour le traitement des patientes atteintes d'un cancer de l'endomètre MSI-H ou dMMR avancé ou récidivant
Il n'existe pas de comparaison directe de pembrolizumab administré en association au lenvatinib et de pembrolizumab en monothérapie. Les médecins doivent considérer le rapport bénéfice/risque des options de traitement disponibles (pembrolizumab en monothérapie ou pembrolizumab en association au lenvatinib) avant d'instaurer le traitement chez les patientes atteintes d'un cancer de l'endomètre MSI-H ou dMMR avancé ou récidivant.
Utilisation du pembrolizumab dans le traitement adjuvant des patients atteints d'un mélanome
Une tendance à l'augmentation de la fréquence des ef ets indésirables sévères et graves chez les patients ≥ 75 ans a été observée. Les données de sécurité de pembrolizumab dans le traitement adjuvant du mélanome sont limitées chez les patients ≥ 75 ans.
Utilisation du pembrolizumab en association à l'axitinib dans le traitement de première ligne des patients atteints d'un CCR
Lorsque pembrolizumab est administré avec axitinib, des fréquences supérieures à celles attendues pour les augmentations des ALAT et des ASAT de Grades 3 et 4 ont été rapportées chez les patients atteints d'un CCR avancé (voir rubrique Effets indésirables). Les enzymes hépatiques doivent être surveillées avant l'initiation du traitement et régulièrement tout au long de celui-ci. Un suivi des enzymes hépatiques plus fréquent que celui effectué lorsque les médicaments sont utilisés en monothérapie peut être envisagé. Les recommandations de prise en charge médicale pour les deux médicaments doivent être suivies (voir rubrique Posologie et mode d'administration et se référer au RCP de l'axitinib).
Utilisation de pembrolizumab en traitement de première ligne des patients atteints de cancer colorectal MSI- H/dMMR
Dans l'étude KEYNOTE-177, le taux de risque pour l'occurrence des évènements de survie globale a été plus élevé pour pembrolizumab en comparaison avec la chimiothérapie pendant les 4 premiers mois de traitement, suivi d'un bénéfice de survie à long terme pour pembrolizumab (voir rubrique Propriétés pharmacodynamiques).
Patients exclus des études cliniques
Les patients présentant les pathologies suivantes ont été exclus des études cliniques : métastases cérébrales actives ; statut de performance ECOG ≥ 2 (à l'exception du carcinome urothélial et du CCR) ; infection par le VIH, le virus de l'hépatite B ou de l'hépatite C ; maladie auto-immune systémique active ; pneumopathie interstitielle diffuse ; pneumopathie inflammatoire antérieure nécessitant un traitement corticoïde systémique ; antécédent d'hypersensibilité sévère à un autre anticorps monoclonal ; patients recevant un traitement immunosuppresseur ; antécédent d'effets indésirables sévères d'origine immunologique avec ipilimumab, définis comme toute toxicité de Grade 4 ou de Grade 3 nécessitant une corticothérapie (> 10 mg/jour de prednisone ou équivalent) pendant plus de 12 semaines. Les patients atteints d'infections actives ont été exclus des études cliniques et leur infection devait avoir été traitée avant de recevoir pembrolizumab. Les patients pour lesquels des infections actives sont apparues durant le traitement par pembrolizumab ont été pris en charge par un traitement médical approprié. Les patients atteints au début dutraitement d'anomalies cliniquement signi ficatives rénale (créatinine > 1,5 fois la LSN) ou hépatique (bilirubine > 1,5 fois la LSN, ALAT/ASAT > 2,5 fois la LSN en l'absence de métastases hépatiques) ont été exclus des études cliniques ; l'information est donc limitée aux patients atteints d'insuffisance rénale sévère et d'insuffisance hépatique modérée à sévère.
Les données sur la sécurité et l'efficacité de KEYTRUDA chez les patients atteints d'un mélanome oculaire sont limitées (voir rubrique Propriétés pharmacodynamiques).
Après un examen attentif du risque potentiellement accru, pembrolizumab peut être utilisé avec une prise en charge médicale appropriée chez ces patients.
Carte de signalement patient
Tous les prescripteurs de KEYTRUDA doivent connaître le document contenant les informations destinées aux médecins et les recommandations de prise en charge. Le prescripteur doit discuter avec le patient des risques du traitement par KEYTRUDA. La carte de signalement patient sera remise au patient avec chaque prescription.
Résumé du profil de sécurité
Pembrolizumab est le plus fréquemment associé à des ef ets indésirables d'origine immunologique. La plupart d'entre eux, y compris les réactions sévères, se sont résolus après initiation d'un traitement médicalapproprié ou arrêt de pembrolizumab (voir « Description d'une sélection d'ef ets indésirables » ci-dessous). Les fréquences mentionnées ci-dessous et dans le tableau 2 sont basées sur tous les effets indésirables rapportés, quelle que soit l'évaluation de la causalité par l'investigateur.
Pembrolizumab en monothérapie (voir rubrique Posologie et mode d'administration)
La sécurité de pembrolizumab en monothérapie a été évaluée dans des études cliniques chez 7 631 patients dans différents types de tumeurs et avec quatre doses (2 mg/kg de poids corporel toutes les 3 semaines, 200 mg toutes les 3 semaines ou 10 mg/kg de poids corporel toutes les 2 ou 3 semaines). Dans cettepopulation de patients, la durée d'observation médiane était de 8,5 mois (de 1 jour à 39 mois) et les effets indésirables les plus fréquents avec pembrolizumab étaient : fatigue (31 %), diarrhée (22 %) et nausée (20 %). La majorité des ef ets indésirables rapportés en monothérapie étaient d'une sévérité de Grades 1 ou 2. Les ef ets indésirables les plus graves étaient des effets indésirables d'origine immunologique et des réactions sévères liées à la perfusion (voir rubrique Mises en garde spéciales et précautions d'emploi). Les incidences des effets indésirables d'origine immunologique étaient de 36,1 %, tous Grades, et 8,9 % pour les Grades 3-5 avec le pembrolizumab en monothérapie au stade adjuvant (n = 1 480), et 24,2 %, tous Grades, et 6,4 % pour lesGrades 3-5 au stade métastatique (n = 5 375). Aucun nouvel effet indésirable d'origine immunologique n'a été identifié au stade adjuvant.
Pembrolizumab en association à une chimiothérapie (voir rubrique Posologie et mode d'administration)
Lorsque pembrolizumab est administré en association, reportez-vous au RCP des médicaments respectifs du traitement en association avant l'initiation du traitement.
La sécurité de pembrolizumab en association à une chimiothérapie a été évaluée dans des études cliniques chez 3 123 patients dans différents types de tumeurs recevant 200 mg, 2 mg/kg de poids corporel ou 10 mg/kg de poids corporel de pembrolizumab toutes les 3 semaines. Dans cette population de patients, les effets indésirables les plus fréquents étaient : anémie (55 %), nausées (54 %), fatigue (38 %),neutropénie (36 %), constipation (35 %), alopécie (35 %), diarrhée (34 %), vomissements (28 %) et diminution de l'appétit (27 %). Les incidences des effets indésirables de Grades 3-5 chez les patients avec un CBNPC étaient de 67 % pour le traitement par pembrolizumab en association et de 66 % pour lachimiothérapie seule, chez les patients avec un CETEC étaient de 85 % pour le traitement par pembrolizumab en association et de 84 % pour la chimiothérapie avec cétuximab, chez les patients atteints d'un cancer de l'œsophage étaient de 86 % pour le traitement par pembrolizumab en association et de 83 % pour la chimiothérapie seule, chez les patients atteints d'un CSTN étaient de 80 % pour le traitement par pembrolizumab en association et de 77 % pour la chimiothérapie seule, et chez les patientes atteintes d'un cancer du col de l'utérus étaient de 82 % pour le traitement par pembrolizumab en association et de 75 % pour la chimiothérapie seule.
Pembrolizumab en association à un inhibiteur de tyrosine kinase (ITK) (voir rubrique Posologie et mode d'administration)
Lorsque pembrolizumab est administré en association à l'axitinib ou au lenvatinib, reportez-vous au RCP de l'axitinib ou du lenvatinib avant l'initiation du traitement. Pour des informations complémentaires sur la sécurité du lenvatinib pour le carcinome à cellules rénales (CCR) avancé, voir le RCP de Kisplyx et pour le cancer de l'endomètre (CE) avancé, voir le RCP de Lenvima. Pour des informations complémentaires sur la sécurité de l'axitinib en cas d'élévation des enzymes hépatiques, voir également la rubrique Mises en garde spéciales et précautions d'emploi.
La
sécurité de pembrolizumab en association à l'axitinib ou au lenvatinib
dans le CCR avancé, et en association au lenvatinib dans le CE avancé,
a été évaluée chez un total de 1 456 patients atteints d'un CCR avancé ou d'un CE avancé recevant 200 mg de pembrolizumab toutes les 3 semaines avec soit 5 mg d'axitinib deux fois par jour, soit 20 mg de lenvatinib une fois par jour dans les études cliniques, selon le cas.
Dans ces populations de patients, les effets indésirables les plus
fréquents étaient : diarrhée (58 %), hypertension (54 %), hypothyroïdie
(46 %), fatigue (41 %), diminution de l'appétit (40 %), nausées (40 %),
arthralgie (30 %), vomissements (28 %), perte de poids (28 %),
dysphonie (28 %), douleurs abdominales (28 %), protéinurie (27 %),
syndrome main-pied (26 %), éruption cutanée (26 %), stomatite (25 %),
constipation (25 %), douleurs musculosquelettiques (23 %), céphalées
(23 %) et toux (21 %). La fréquence des effets indésirables de Grade
3-5 chez les patients atteints de CCR était de 80 % pour le
pembrolizumab en association à l'axitinib ou au lenvatinib et de 71 %
pour le sunitinib seul. Chez les patients atteints de CE, la fréquence
des effets indésirables de Grade 3-5 était de 89 % pour le
pembrolizumab en association au lenvatinib et de 73 % pour la
chimiothérapie seule.
Résumé tabulé des effets indésirables
Les
effets indésirables observés dans les études cliniques avec
pembrolizumab en monothérapie ou en association avec la chimiothérapie ou d'autres médicaments anti-cancéreux,
ou rapportés depuis la commercialisation de pembrolizumab sont listés
dans le tableau 2. Ces effets sont présentés par classes de systèmes d'organes et par fréquence. Les fréquences sont définies ainsi : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000) et
fréquence indéterminée (ne peut être estimée sur la base des données
disponibles). Pour chaque groupe de fréquence, les effets indésirables
sont présentés par ordre décroissant de gravité. Les effets
indésirables connus pour survenir avec pembrolizumab ou les médicaments
du traitement en association administrés seuls peuvent apparaître
pendant le traitement avec ces médicaments en association, même si ces
effets n'ont pasété rapportés au cours des études cliniques avec l'association thérapeutique.
Pour plus d'informations sur la sécurité lorsque pembrolizumab est
administré en association, reportez-vous au RCP des médicaments
respectifs du traitement en association.
Tableau 2 : Effets indésirables chez les patients traités par pembrolizumab†
| Monothérapie | En association avec une chimiothérapie | En association avec axitinib ou lenvatinib | |
| Infections et infestations | |||
| Très fréquent | Infections des voies urinaires | ||
| Fréquent | pneumonie | pneumonie | pneumonie |
| Affections hématologiques et du système lymphatique | |||
| Très fréquent | anémie | neutropénie, anémie, thrombopénie, leucopénie | anémie |
| Fréquent | thrombopénie, neutropénie, lymphopénie | neutropénie fébrile, lymphopénie | neutropénie, thrombopénie, lymphopénie, leucopénie |
| Peu fréquent | leucopénie, thrombopénie immunitaire,éosinophilie | éosinophilie | éosinophilie |
| Rare | lymphohistiocytose hémophagocytaire, anémie hémolytique, érythroblastopénie | anémie hémolytique, thrombopénie immunitaire | |
| Affections du système immunitaire | |||
| Fréquent | réaction liée à la perfusion* | réaction liée à la perfusion* | réaction liée à la perfusion* |
| Peu fréquent | sarcoïdose* | ||
| Rare | sarcoïdose | ||
| Fréquence indéterminée | rejet de greffe d'organe solide | ||
| Affections endocriniennes | |||
| Très fréquent | hypothyroïdie* | hypothyroïdie* | hypothyroïdie |
| Fréquent | hyperthyroïdie | insuffisance surrénalienne*, thyroïdite*, hyperthyroïdie* | insuffisance surrénalienne*, hyperthyroïdie, thyroïdite* |
| Peu fréquent | insuffisance surrénalienne*, hypophysite*, thyroïdite* | hypophysite* | hypophysite* |
| Rare | hypoparathyroïdie | hypoparathyroïdie | hypoparathyroïdie |
| Troubles du métabolisme et de la nutrition | |||
| Très fréquent | diminution de l'appétit | hypokaliémie, diminution del'appétit | diminution de l'appétit |
| Fréquent | hyponatrémie, hypokaliémie, hypocalcémie | hyponatrémie, hypocalcémie | hyponatrémie, hypokaliémie, hypocalcémie |
| Peu fréquent | diabète de type I* | diabète de type I* | diabète de type I* |
| Monothérapie | En association avec une chimiothérapie | En association avec axitinib ou lenvatinib | |
| Affections psychiatriques | |||
| Très fréquent | insomnie | ||
| Fréquent | insomnie | insomnie | |
| Affections du système nerveux | |||
| Très fréquent | céphalée | neuropathie périphérique, céphalées, étourdissements, dysgueusie | céphalée, dysgueusie |
| Fréquent | étourdissements, neuropathie périphérique, léthargie, dysgueusie | léthargie | étourdissements, neuropathie périphérique, léthargie |
| Peu fréquent | syndrome myasthénique*, épilepsie | encéphalite*, épilepsie | syndrome myasthénique*, encéphalite* |
| Rare | syndrome de Guillain-Barré*, encéphalite*, myélite*, névriteoptique, méningite (aseptique) * | syndrome de Guillain-Barré*,syndrome myasthénique | névrite optique |
| Affections oculaires | |||
| Fréquent | sécheresse oculaire | sécheresse oculaire | sécheresse oculaire |
| Peu fréquent | uvéite* | uvéite? | |
| Rare | syndrome de Vogt-Koyanagi-Harada | uvéite* | syndrome de Vogt-Koyanagi-Harada |
| Affections cardiaques | |||
| Fréquent | arythmie cardiaque‡ (y compris fibrillation auriculaire) | arythmie cardiaque‡ (ycompris fibrillation auriculaire) | arythmie cardiaque‡ ( y compris fibrillation auriculaire) |
| Peu fréquent | myocardite, épanchement péricardique,péricardite | myocardite*, épanchement péricardique, péricardite | myocardite, épanchement péricardique |
| Affections vasculaires | |||
| Très fréquent | hypertension | ||
| Fréquent | hypertension | hypertension | |
| Peu fréquent | vascularite* | vascularite* | |
| Rare | vascularite* | ||
| Monothérapie | En association avec une chimiothérapie | En association avec axitinib ou lenvatinib | |
| Affections respiratoires, thoraciques et médiastinales | |||
| Très fréquent | dyspnée, toux | dyspnée, toux | dyspnée, toux |
| Fréquent | pneumopathie inflammatoire* | pneumopathie inflammatoire* | pneumopathie inflammatoire* |
| Affections gastro-intestinales | |||
| Très fréquent | diarrhée, douleurs abdominales*, nausées, vomissements, constipation | nausées, diarrhée, vomissements, douleurs abdominales*,constipation | diarrhée, douleurs abdominales*, nausées, vomissements, constipation |
| Fréquent | colite*, sécheresse buccale | colite*, gastrite,sécheresse buccale | colite*, pancréatite*, gastrite, sécheresse buccale |
| Peu fréquent | pancréatite*, gastrite, ulcération gastro-intestinale* | pancréatite*, ulcération gastro- intestinale* | ulcération gastro- intestinale* |
| Rare | perforation de l'intestin grêle | perforation de l'intestin grêle | perforation de l'intestin grêle |
| Troubles hépatobiliaires | |||
| Fréquent | hépatite* | hépatite* | hépatite* |
| Rare | cholangite sclérosante | cholangite sclérosante* | |
| Affections de la peau et du tissu sous-cutané | |||
| Très fréquent | prurit*, éruption cutanée* | alopécie, éruptioncutanée*, prurit* | éruption cutanée*, prurit* |
| Fréquent | réactions cutanées sévères*, érythème, dermatite, sécheresse cutanée, vitiligo*, eczéma, alopécie, dermatite acnéiforme | réactions cutanées sévères*, érythème, dermatite acnéiforme, dermatite, sécheresse cutanée, eczéma | réactions cutanées sévères*, dermatite, sécheresse cutanée, érythème, dermatite acnéiforme, alopécie |
| Peu fréquent | psoriasis, kératose lichénoïde*, papule, modification de la couleur des cheveux | psoriasis, kératose lichénoïde*, vitiligo*, papule | eczéma, kératose lichénoïde*, psoriasis, vitiligo*, papule, modification de la couleur descheveux |
| Rare | syndrome de Stevens-Johnson, érythème noueux, nécrolyse épidermique toxique | syndrome de Stevens-Johnson, érythème noueux, modification de la couleur des cheveux | nécrolyse épidermique toxique, syndrome de Stevens-Johnson |
| Monothérapie | En association avec une chimiothérapie | En association avec axitinib ou lenvatinib | |
| Affections musculo-squelettiques et systémiques | |||
| Très fréquent | douleur musculo-squelettique*, arthralgie | arthralgie, douleurmusculo- squelettique*, myosite* | arthralgie, douleur musculo-squelettique*, myosite*, douleuraux extrémités |
| Fréquent | myosite*, douleuraux extrémités, arthrite* | douleur aux extrémités, arthrite* | arthrite* |
| Peu fréquent | ténosynovite* | ténosynovite* | ténosynovite* |
| Rare | syndrome de Sjögren | syndrome de Sjögren | syndrome de Sjögren |
| Troubles du rein et des voies urinaires | |||
| Fréquent | insuffisance rénaleaiguë | néphrite* | |
| Peu fréquent | néphrite* | néphrite*, cystite non infectieuse | |
| Rare | cystite noninfectieuse | cystite non infectieuse | |
| Troubles généraux et anomalies au site d'administration | |||
| Très fréquent | fatigue, asthénie,œdème*, fièvre | fatigue, asthénie,fièvre, œdème* | fatigue, asthénie,œdème*, fièvre |
| Fréquent | syndrome pseudo-grippal, frissons | syndrome pseudo-grippal, frissons | syndrome pseudo-grippal, frissons |
| Investigations | |||
| Très fréquent | augmentation de l'alanine aminotransférase, augmentation de l'aspartate aminotransférase | augmentation de la lipase, augmentation de l'alanine aminotransférase, augmentation de l'aspartate aminotransférase, augmentation de la créatininémie | |
| Fréquent | augmentation de l'alanine aminotransférase, augmentation de l'aspartate aminotransférase, augmentation des phosphatases alcalines sanguines, hypercalcémie, augmentation de la bilirubinémie, augmentation de la créatininémie | augmentation de la créatininémie, augmentation des phosphatases alcalines sanguines, hypercalcémie, augmentation de la bilirubinémie | augmentation de l'amylase,augmentation de la bilirubinémie, augmentation des phosphatases alcalines sanguines, hypercalcémie |
| Peu fréquent | augmentation de l'amylase | augmentation de l'amylase | |
‡ Sur la base d'une requête standard incluant bradyarythmie et tachyarythmie.
*Les termes suivants représentent un groupe d'évènements liés qui décrivent un état pathologique plutôt qu'un évènement isolé :
- réaction liée à la perfusion (hypersensibilité médicamenteuse, réaction anaphylactique, réaction anaphylactoïde, hypersensibilité, réaction d'hypersensibilité liée à la perfusion, syndrome de relargage des cytokines et maladie sérique)
- sarcoïdose (sarcoïdose cutanée et sarcoïdose pulmonaire)
- hypothyroïdie (myxœdème, hypothyroïdie à médiation immunitaire et hypothyroïdie auto-immune)
- insuf isance surrénalienne (maladie d'Addison, insuf isance corticosurrénalienne aiguë et insuffisance cortico- surrénalienne secondaire)
- thyroïdite (thyroïdite auto-immune, troubles thyroïdiens, thyroïdite aiguë et thyroïdite à médiation immunitaire)
- hyperthyroïdie (maladie de Basedow)
- hypophysite (hypopituitarisme et hypophysite lymphocytaire)
- diabète de type 1 (acidocétose diabétique)
- syndrome myasthénique (myasthénie grave, y compris exacerbation)
- encéphalite (encéphalite auto-immune et encéphalite non infectieuse)
- syndrome de Guillain-Barré (neuropathie axonale et polyneuropathie démyélinisante)
- myélite (y compris myélite transverse)
- méningite aseptique (méningite et méningite non infectieuse)
- uvéite (choriorétinite, iritis et iridocyclite)
- myocardite (myocardite auto-immune)
- vascularite (vascularite du système nerveux central, aortite et artérite à cellules géantes)
- pneumopathie inflammatoire (pneumopathie interstitielle diffuse, pneumopathie organisée, pneumopathie à médiation immunitaire et maladie pulmonaire à médiation immunitaire)
- douleur abdominale (gêne abdominale, douleur abdominale haute et douleur abdominale basse)
- colite (colite microscopique, entérocolite, entérocolite hémorragique, colite auto-immune et entérocolite à médiation immunitaire)
- pancréatite (pancréatite auto-immune, pancréatite aiguë et pancréatite à médiation immunitaire)
- ulcération gastro-intestinale (ulcère gastrique et ulcère duodénal)
- hépatite (hépatite auto-immune, hépatite à médiation immunitaire, atteinte hépatique d'origine médicamenteuse et hépatite aiguë)
- cholangite sclérosante (cholangite à médiation immunitaire)
- prurit (urticaire, urticaire papuleuse et prurit génital)
- éruption cutanée (éruption cutanée érythémateuse, éruption folliculaire, éruption maculaire, éruption maculo-papuleuse, éruption papuleuse, éruption pruritigineuse, éruption vésiculaire et rash génital)
- réactions cutanées sévères (rash exfoliatif, pemphigus, et évènements suivants de Grade ≥ 3
: dermatite bulleuse, dermatite exfoliative, dermatite exfoliative
généralisée, érythème polymorphe, lichen plan, lichen plan buccal,
pemphigoïde, prurit, prurit génital, éruption cutanée, éruption cutanée érythémateuse, éruption maculo-papuleuse, éruption cutanée prurigineuse, éruption pustuleuse, nécrose cutanée et éruption cutanée toxique) - vitiligo (dépigmentation cutanée, hypopigmentation cutanée et hypopigmentation de la paupière)
- kératose lichénoïde (lichen plan et lichen scléreux)
- douleur musculo-squelettique (gêne musculo-squelettique, douleur dorsale, raideur musculo-squelettique, douleur thoracique musculo-squelettique et torticolis)
- myosite (myalgie, myopathie, myosite nécrosante, pseudo-polyarthrite rhizomélique et rhabdomyolyse)
- arthrite (gonflement des articulations, polyarthrite, épanchement articulaire, arthrite auto-immune et arthrite à médiation immunitaire)
- ténosynovite (tendinite, synovite et douleur aux tendons)
- néphrite (néphrite auto-immune, néphrite tubulo-interstitielle et insuffisance rénale, insuffisance rénale aiguë ou atteinte rénale aiguë avec néphrite avérée, syndrome néphrotique, glomérulonéphrite, glomérulonéphrite membraneuse et glomérulonéphrite aiguë)
- œdème (œdème périphérique, œdème généralisé, surcharge liquidienne, rétention liquidienne, œdème palpébral et œdème labial, œdème du visage, œdème localisé et œdème périorbitaire)
Description d'une sélection d'effets indésirables
Les données concernant les ef ets indésirables d'origine immunologique suivants sont basées sur les patients ayant reçu pembrolizumab selon quatre posologies (2 mg/kg de poids corporel toutes les 3 semaines, 10 mg/kg de poids corporel toutes les 2 ou 3 semaines ou 200 mg toutes les 3 semaines) dans les étudescliniques (voir rubrique Propriétés pharmacodynamiques). Les recommandations de prise en charge de ces effets indésirables sont décrites en rubrique Mises en garde spéciales et précautions d'emploi.
Effets indésirables d'origine immunologique (voir rubrique Mises en garde spéciales et précautions d'emploi)
Pneumopathie inflammatoire d'origine immunologique
Une
pneumopathie inflammatoire est survenue chez 324 (4,2 %) patients
recevant pembrolizumab, y compris des cas de Grade 2, 3, 4 ou 5 chez
143 (1,9 %), 81 (1,1 %), 19 (0,2 %) et 9 (0,1 %) patients,
respectivement.
Le délai d'apparition médian d'une pneumopathie inflammatoire a été de 3,9 mois (de 2 jours à 27,2 mois) et la durée médiane a été de 2,0 mois (de 1 jour à 51,0+ mois). La pneumopathie inflammatoire était plus fréquente chez les patients ayant des antécédents d'irradiation thoracique antérieure (8,1 %) que chez les patients n'ayant pas reçu d'irradiation thoracique préalable (3,9 %). Une pneumopathie inflammatoire a conduit à un arrêt de pembrolizumab chez 131 (1,7 %) patients. La pneumopathie inflammatoire s'est résolue chez 196 patients, 6 avec des séquelles.
Pour les patients atteints de CBNPC, une pneumopathie inflammatoire est survenue chez 160 (5,7 %), y compris des cas de Grade 2, 3, 4 ou 5 chez 62 (2,2 %), 47 (1,7 %), 14 (0,5 %) et 10 (0,4 %) patients, respectivement. Chez les patients atteints de CBNPC, une pneumopathie inflammatoire est survenue chez 8,9 % patients ayant des antécédents d'irradiation thoracique antérieure. Chez les patients atteints de LHc, l'incidence de la pneumopathie inflammatoire (tous grades) variait entre 5,2 % et 10,8 % pour les patients avec un LHc dans l'étude KEYNOTE-087 (n = 210) et l'étude KEYNOTE-204 (n = 148), respectivement.
Colite d'origine immunologique
Une colite est survenue chez 158 (2,1 %) patients recevant pembrolizumab, y compris des cas de Grade 2, 3 ou 4 chez 49 (0,6 %), 82 (1,1 %) et 6 (0,1 %) patients, respectivement. Le délai d'apparition médian de la colite a été de 4,3 mois (de 2 jours à 24,3 mois) et la durée médiane a été de 1,1 mois (de 1 jour à 45,2 mois). Une colite a conduit à un arrêt de pembrolizumab chez 48 (0,6 %) patients. La colite s'est résolue chez 132 patients, 2 avec des séquelles. Chez les patients atteints de cancer colorectal traités par pembrolizumab en monothérapie (n = 153), l'incidence de la colite était de 6,5 % (tous grades) avec 2,0 % de Grade 3 et 1,3 % de Grade 4.
Hépatite d'origine immunologique
Une hépatite est survenue chez 80 (1,0 %) patients recevant pembrolizumab, y compris des cas de Grade 2, 3 ou 4 chez 12 (0,2 %), 55 (0,7 %) et 8 (0,1 %) patients, respectivement. Le délai d'apparition médian de l'hépatite a été de 3,5 mois (de 8 jours à 26,3 mois) et la durée médiane a été de 1,3 mois (de 1 jour à 29,0+ mois). Une hépatite a conduit à un arrêt de pembrolizumab chez 37 (0,5 %) patients. L'hépatite s'est résolue chez 60 patients.
Néphrite d'origine immunologique
Une néphrite est survenue chez 37 (0,5 %) patients recevant pembrolizumab en monothérapie, y compris des cas de Grade 2, 3 ou 4 chez 11 (0,1 %), 19 (0,2 %) et 2 (< 0,1 %) patients, respectivement. Le délaid'appa rition médian de la néphrite a été de 4,2 mois (de 12 jours à 21,4 mois) et la durée médiane a été de 3,3 mois (de 6 jours à 28,2+ mois). Une néphrite a conduit à un arrêt de pembrolizumab chez 17 (0,2 %) patients. La néphrite s'est résolue chez 25 patients, 5 avec des séquelles. Chez les patients atteints d'un CBNPC non-épidermoïde traités par pembrolizumab associé à une chimiothérapie pemetrexed et sel de platine (n = 488), l'incidence de la néphrite était de 1,4 % (tous grades) avec 0,8 % de Grade 3 et 0,4 % de Grade 4.
Endocrinopathies d'origine immunologique
Une insuffisance surrénalienne est survenue chez 74 (1,0 %) patients, y compris des cas de Grade 2, 3 et 4 chez 34 (0,4 %), 31 (0,4 %) et 4 (0,1 %) patients recevant pembrolizumab, respectivement. Le délaid'apparition médian de l'insuffisance surrénalienne a été de 5, 4 mois (de 1 jour à 23,7 mois) et la durée médiane n'a pas été atteinte (de 3 jours à 40,1+ mois). Une insuffisance surrénalienne a conduit à un arrêt de pembrolizumab chez 13 (0,2 %) patients. L'insuffisance surrénalienne s'est résolue chez 28 patients, 11 avec des séquelles.
Une
hypophysite est survenue chez 52 (0,7 %) patients recevant
pembrolizumab, y compris des cas de Grade 2, 3 ou 4 chez 23 (0,3 %), 24
(0,3 %) et 1 (< 0,1 %) patients, respectivement. Le délai d'apparition médian de l'hypophysite a été de 5,9
mois (de 1 jour à 17,7 mois) et la durée médiane a été de 3,6 mois (de
3 jours à 48,1+ mois). Une hypophysite a conduit à un arrêt de
pembrolizumab chez 14 (0,2 %) patients.
L'hypophysite s'est résolue chez 23 patients, 8 avec des séquelles.
Une hyperthyroïdie est survenue chez 394 (5,2 %) patients recevant pembrolizumab, y compris des cas de Grade 2 ou 3 chez 108 (1,4 %) et 9 (0,1 %) patients, respectivement. Le délai d'apparition médian de l'hyperthyroïdie a été de 1,4 mois (de 1 jour à 23,2 mois) et la durée médiane a été de 1,6 mois (de 4 jours à 43,1+ mois). Une hyperthyroïdie a conduit à un arrêt de pembrolizumab chez 4 (0,1 %) patients. L'hyperthyroïdie s'est résolue chez 326 (82,7 %) patients, 11 avec des séquelles. Chez les patients atteints d'un CCR et d'un mélanome traités par pembrolizumab en monothérapie au stade adjuvant (n = 1 480), l'incidence de l'hyperthyroïdie était de 10,9 %, dont la majorité était de Grade 1 ou 2.
Une
hypothyroïdie est survenue chez 939 (12,3 %) patients recevant
pembrolizumab, y compris des cas de Grade 2 ou 3 chez 687 (9,0 %) et 8
(0,1 %) patients, respectivement. Le délai d'apparition médian de l'hypothyroïdie a été de 3,4 mois (de 1 jour à 25,9 mois) et la durée médiane n'a pas été atteinte (de 2 jours à 63,0+ mois). L'hypothyroïdie a conduit à un arrêt de pembrolizumab chez 6 (0,1 %) patients.
L'hypothyroïdie s'est résolue chez 216 (23,0 %) patients, 16 avec des séquelles. Chez les patients atteints d'un LHc (n = 389), l'incidence de l'hypothyroïdie était de 17 %, toutes étant de Grade 1 ou 2. Chez les patients atteints d'un CETEC traités par pembrolizumab en monothérapie (n = 909), l'incidence de l'hypothyroïdie était de 16,1 % (tous grades) avec 0,3 % de Grade 3. Chez les patients atteints d'un CETEC traités par pembrolizumab en association à une chimiothérapie à base de sels de platine et de 5-FU (n = 276), l'incidence de l'hypothyroïdie était de 15,2 %, toutes étant de Grade 1 ou 2. Chez les patients traités par pembrolizumab en association à l'axitinib ou au lenvatinib (n=1 456), l'incidence de l'hypothyroïdie était de 46,2 % (tous Grades) avec 0,8 % de Grade 3 ou 4. Chez les patients atteints d'un CCR et d'un mélanome traités
par pembrolizumab en monothérapie au stade adjuvant (n = 1 480),
l'incidence de l'hypothyroïdie était de 17,7 %, dont la majorité était
de Grade 1 ou 2.
Efets indésirables cutanés d'origine immunologique
Des réactions cutanées sévères d'origine immunologique sont survenues chez 130 (1,7 %) patients recevant pembrolizumab, y compris des cas de Grade 2, 3, 4 ou 5 chez 11 (0,1 %), 103 (1,3 %), 1 (< 0,1 %) et 1 (< 0,1%) patients, respectivement. Le délai d'apparition médian des réactions cutanées sévères a été de 2,8 mois (de 2 jours à 25,5 mois). La durée médiane a été de 1,9 mois (de 1 jour à 47,1+ mois). Des réactions cutanées sévères ont conduit à un arrêt de pembrolizumab chez 18 (0,2 %) patients. Les réactions cutanées sévères se sont résolues chez 95 patients, 2 avec des séquelles.
De rares cas de SSJ et de NET, dont certains d'issue fatale, ont été observés (voir rubriques Posologie et mode d'administration et Mises en garde spéciales et précautions d'emploi).
Complications d'une GCSHallogénique dans le LHc
Parmi les 14 patients de KEYNOTE-013 ayant reçu une GCSH allogénique après un traitement par pembrolizumab, 6 patients ont développé une GVH aiguë et 1 patient a développé une GVH chronique, dont aucune n'a été fatale. Deux patients ont présenté une MVO hépatique, dont une d'issue fatale. Un patient a présenté un syndrome de prise de greffe après transplantation.
Parmi les 32 patients de KEYNOTE-087 ayant reçu une GCSH allogénique après un traitement par pembrolizumab, 16 patients ont développé une GVH aiguë et 7 patients ont développé une GVH chronique, dont deux d'issue fatale. Aucun patient n'a présenté de MVO hépatique. Aucun patient n'a présenté de syndrome de prise de greffe après transplantation.
Parmi les 14 patients de KEYNOTE-204 ayant reçu une GCSH allogénique après un traitement par pembrolizumab, 8 patients ont développé une GVH aiguë et 3 patients ont développé une GVH chronique, aucune d'issue fatale. Aucun patient n'a présenté de MVO hépatique. Un patient a présenté un syndrome de prise de greffe après transplantation.
Enzymes hépatiques élevées lorsque pembrolizumab est associé à l'axitinib dans le CCR
Dans une étude clinique chez des patients atteints d'un CCR non préalablement traité recevant pembrolizumab en association à l'axitinib, une augmentation des ALAT (20 %) et des ASAT (13 %) de Grades 3 et 4 a été observée avec une incidence plus élevée qu'attendue. La durée médiane d'apparition de l'augmentation des ALAT était de 2,3 mois (de 7 jours à 19,8 mois). Chez les patients avec des ALAT ≥ 3 fois la LSN (Grades 2-4, n = 116), l'augmentation des ALAT s'est améliorée jusqu'aux Grades 0-1 chez 94 % d'entre eux. Cinquante-neuf pour cent des patients présentant des ALAT augmentées ont reçu des corticostéroïdes systémiques. Parmi les patients qui se sont rétablis, une réintroduction a été faite chez92 (84 %) d'entre eux avec soit pembrolizumab (3 %) soit axitinib (31 %) en monothérapie soit les deux (50 %). Parmi ces patients, 55 % n'ont pas eu de réapparition des ALAT > 3 fois la LSN, et parmi les patients ayant présenté une réapparition des ALAT > 3 fois la LSN, tous se sont rétablis. Il n'y a pas eu d'ef ets indésirables hépatiques de Grade 5.
Anomalies des valeurs biologiques
Chez les patients traités par pembrolizumab en monothérapie, la proportion de patients ayant présenté une variation des paramètres biologiques vers des anomalies de Grade 3 ou 4 par rapport aux valeurs à l'inclusion a été la suivante : 9,4 % pour une diminution des lymphocytes, 7,4 % pour une diminution du sodium, 5,8 %pour une diminution de l'hémoglobine, 5,3 % pour une diminution du phosphate, 5,3 % pour une augmentation du glucose, 3,3 % pour une augmentation des ALAT, 3,1 % pour une augmentation des ASAT, 2,6 % pour une augmentation des phosphatases alcalines, 2,3 % pour une diminution du potassium, 2,1 % pour une augmentation du potassium, 1,9 % pour une diminution des neutrophiles, 1,8 % pour une diminution des plaquettes, 1,8 % pour une augmentation du calcium, 1,7 % pour une augmentation de labilirubine, 1,5 % pour une diminution du calcium, 1,4 % pour une diminution de l'albumine, 1,3 % pour une augmentation de la créatinine, 1,2 % pour une diminution du glucose, 0,8 % pour une diminution des leucocytes, 0,7 % pour une augmentation du magnésium, 0,5 % pour une augmentation du sodium, 0,4 % pour une augmentation de l'hémoglobine et 0,2 % pour une diminution du magnésium.
Chez les patients traités par pembrolizumab en association à la chimiothérapie, la proportion de patients ayant présenté une variation des paramètres biologiques vers des anomalies de Grade 3 ou 4 par rapport aux valeurs à l'inclusion a été la suivante : 44,0 % pour une diminution des neutrophiles, 29,4 % pour une diminution des leucocytes, 26,9 % pour une diminution des lymphocytes, 22,1 % pour une diminution del'hémoglobine, 13,2 % pour une diminution des plaquettes, 11,0 % pour une diminution du sodium, 7,7 % pour une diminution du phosphate, 6,8 % pour une augmentation des ALAT, 6,8 % pour une diminution du potassium, 6,1 % pour une augmentation du glucose, 5,6 % pour une augmentation des ASAT, 3,5 % pour une diminution du calcium, 3,2 % pour une augmentation du potassium, 2,9 % pour une augmentation de lacréatinine, 2,2 % pour une diminution de l'albumine, 2,1 % pour une augmentation des phosphatases alcalines, 2,0 % pour une augmentation de la bilirubine, 2,0 % pour une augmentation du calcium, 1,3 % pour une augmentation de l'INR (prothrombine), 1,2 % pour une diminution du glucose et 0,5 % pour une augmentation du sodium.
Chez les patients traités par pembrolizumab en association à l'axitinib ou au lenvatinib, la proportion de patients ayant présenté une variation des paramètres biologiques vers des anomalies de Grade 3 ou 4 par rapport aux valeurs à l'inclusion a été la suivante : 23,0 % pour une augmentation de la lipase (non mesurée chez les patients traités par pembrolizumab et axitinib), 12,0 % pour une diminution des lymphocytes, 11,4 % pour une diminution du sodium, 11,2 % pour une augmentation de l'amylase, 11,2 % pour une augmentation des triglycérides, 10,4 % pour une augmentation des ALAT, 8,9 % pour une augmentation des ASAT, 7,8 % pour une augmentation du glucose, 6,8 % pour une diminution du phosphate, 6,1 % pour une diminution du potassium, 5,1 % pour une augmentation du potassium, 4,5 % pour une augmentation du cholestérol, 4,4 % pour une augmentation de la créatinine, 4,2 % pour une diminution de l'hémoglobine, 4,0 % pour une diminution du magnésium, 3,5 % pour une diminution des neutrophiles, 3,1 % pour une augmentation des phosphatases alcalines, 3,0 % pour une diminution des plaquettes, 2,8 % pour une augmentation de la bilirubine, 2,2 % pour une diminution du calcium, 1,7 % pour une diminution des globules blancs, 1,6 % pour une augmentation du magnésium, 1,5 % pour une augmentation de l'INR (prothrombine), 1,4 % pour une diminution du glucose, 1,2 % pour une diminution de l'albumine, 1,2 % pour une augmentation du calcium, 0,4 % pour une augmentation du sodium et 0,1 % pour une augmentation de l'hémoglobine.
Immunogénicité
Dans les études cliniques menées chez les patients traités par pembrolizumab en monothérapie à la dose de 2 mg/kg de poids corporel toutes les trois semaines, 200 mg toutes les trois semaines ou 10 mg/kg de poids corporel toutes les 2 ou 3 semaines, 36 (1,8 %) des 2 034 patients évaluables ont été testés positifs pour des anticorps anti-pembrolizumab apparus au cours du traitement dont 9 (0,4 %) patients avec des anticorpsneutralisants contre pembrolizumab. Il n'a pas été mis en évidence de modification du profil pharmacocinétique ou de tolérance en présence d'anticorps anti-pembrolizumab liants ou neutralisants.
Population pédiatrique
La sécurité de pembrolizumab en monothérapie a été évaluée à la dose de 2 mg/kg de poids corporel toutes les 3 semaines dans l'étude de phase I/II KEYNOTE-051 réalisée chez 161 patients pédiatriques âgés de 9 mois à 17 ans atteints d'un mélanome avancé, d'un lymphome ou de tumeurs solides avancées PD-L1 positives, en rechute ou réfractaires. La population avec un LHc (n = 22) incluait des patients âgés de 11 à 17 ans. Le profil de sécurité chez les patients pédiatriques était généralement similaire à celui observé chez les adultes traités par pembrolizumab. Les effets indésirables les plus fréquents (rapportés chez au moins 20 % des patients pédiatriques) étaient : pyrexie (33 %), vomissements (30 %), céphalées (26 %), douleurs abdominales (22 %), anémie (21 %), toux (21 %) et constipation (20 %). La majorité des effets indésirablesrapportés en monothérapie étaient d'une sévérité de Grades 1 ou 2. Soixante-seize (47,2 %) patients présentaient 1 ou plusieurs effets indésirables de Grades 3 à 5 dont 5 (3,1 %) patients avec 1 ou plusieurs effets indésirables ayant entraîné le décès. Les fréquences sont basées sur tous les effets indésirablesrapportés, quelle que soit l'évaluation de la causalité par l'investigateur. Les données de sécurité à long terme du pembrolizumab chez les adolescents atteints de mélanome de stade IIB, IIC et III traités au stade adjuvant sont actuellement indisponibles.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
SURVEILLANCE :
- Respiratoire : rechercher les signes et symptômes de pneumopathie inflammatoire.
- Gastro-intestinal : rechercher les signes et symptômes de colite.
- Hépatique : rechercher les modifications de la fonction hépatique et signes d'hépatite.
- Rénal : rechercher les signes de néphrite.
- Surrénales: rechercher les signes et symptômes d'une insuffisance
surrénalienne et d'une hypophysite (y compris hypopituitarisme).
- Glycémie.
- Fonction thyroïdienne: à l'instauration du traitement, régulièrement
pendant le traitement et en fonction de l'évaluation clinique.
- Réaction cutanées.
En cas de myocardite, d'encéphalite ou de syndrome de Guillain-Barré de Grades 3 ou 4, ARRETER DEFINITIVEMENT le pembrolizumab.
CANCER BRONCHIQUE NON A PETITES CELLULES, CARCINOME UROTHELIAL ou
CARCINOME EPIDERMOIDE DE LA TETE ET DU COU, CANCER DU SEIN TRIPLE
NEGATIF ou CANCER DU COL DE L'UTERUS : Les patients doivent être
sélectionnés par la présence d'une expression tumorale de PD-L1
confirmée par un test validé.
La
sélection des patients pour ce traitement en monothérapie doit être
confirmée par un test validé sur le
statut tumoral MSI-H/dMMR pour les indications de cancer colorectal,
cancer de l'endomètre, cancer gastrique, cancer de l'intestin grêle ou
cancer des voies biliaires.
FEMMES EN AGE DE PROCREER : Elles doivent utiliser une méthode efficace de
contraception pendant le traitement par pembrolizumab et pendant au
moins 4 mois après la dernière administration.
DISCUTER avec le patient des risques du traitement par pembrolizumab et lui REMETTRE la carte de signalement patient.
Femmes en âge de procréer
Les femmes en âge de procréer doivent utiliser une méthode efficace de contraception pendant le traitement par pembrolizumab et pendant au moins 4 mois après la dernière administration de pembrolizumab.
Grossesse
Il n'existe pas de données sur l'utilisation de pembrolizumab chez la femme enceinte. Aucune étude de reproduction n'a été conduite avec pembrolizumab ; cependant, dans des modèles murins de grossesse, le blocage de la voie de signalisation PD-L1 a montré une modification de la tolérance au fœtus et a conduit à une augmentation des pertes fœtales (voir rubrique Données de sécurité préclinique). Sur la base du mécanisme d'action, ces résultats indiquent que l'administration de pembrolizumab pendant la grossesse expose à un risque potentiel d'effet nocif sur le fœtus, incluant des taux plus élevés d'avortement ou d'enfants mort-nés. Il est connu que les immunoglobulines humaines G4 (IgG4) passent la barrière placentaire ; par conséquent pembrolizumab, étant une IgG4, a la possibilité d'être transmis de la mère au fœtus en développement. Pembrolizumab ne doit pas être utilisé pendant la grossesse à moins que l'état clinique de la femme ne nécessite un traitement par pembrolizumab.
Allaitement
On ne sait pas si pembrolizumab est excrété dans le lait maternel. Les anticorps étant connus pour être secrétés dans le lait maternel, un risque pour les nouveau-nés/nourrissons ne peut être exclu. Une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre pembrolizumab, en prenant en compte le bénéfice de l'allaitement pour l'enfant et le bénéfice du traitement par pembrolizumab pour la femme.
Fertilité
Aucune donnée clinique n'est disponible concernant les effets possibles de pembrolizumab sur la fertilité. Les études de toxicité à dose répétée de 1 mois et 6 mois menées sur des singes n'ont pas montré d'effets notables sur les organes reproducteurs mâles et femelles (voir rubrique Données de sécurité préclinique).
Aucune étude pharmacocinétique formelle d'interaction médicamenteuse n'a été réalisée avec pembrolizumab. Pembrolizumab étant éliminé de la circulation par catabolisme, aucune interactionmédicamenteuse métabolique n'est attendue.
L'utilisation de corticostéroïdes ou d'immunosuppresseurs systémiques doit être évitée avant le début du traitement par pembrolizumab, du fait de leur possible interférence avec l'activité pharmacodynamique et l'efficacité de pembrolizumab. Néanmoins, les corticostéroïdes systémiques ou d'autres immunosuppresseurs peuvent être utilisés après l'instauration de pembrolizumab pour traiter des effets indésirables d'origine immunologique (voir rubrique Mises en garde spéciales et précautions d'emploi). Les corticostéroïdes peuvent aussi être utilisés en prémédication quand pembrolizumab est utilisé en association à une chimiothérapie, en prophylaxie antiémétique et/ou pour atténuer les effets indésirables liés à la chimiothérapie.
Le traitement doit être initié et supervisé par des médecins qualifiés et expérimentés dans l'utilisation de traitements anticancéreux.
Test PD-L1
Si cela est spécifié dans l'indication, la sélection des patients pour
le traitement par KEYTRUDA basée sur l'expression tumorale de PD-L1
doit être confirmée par un test validé (voir rubriques Indications thérapeutiques, Mises en garde spéciales et précautions d'emploi, Effets indésirables et Propriétés pharmacodynamiques).
Test MSI/MMR
Si cela est spécifié dans l'indication, la sélection des patients pour
le traitement par KEYTRUDA basée sur le statut tumoral MSI-H/dMMR doit
être confirmée par un test validé (voir rubriques Indications thérapeutiques et Propriétés pharmacodynamiques).
Posologie
La dose recommandée de KEYTRUDA chez les adultes est soit de 200 mg toutes les 3 semaines, soit de 400 mg toutes les 6 semaines, administrée en perfusion intraveineuse pendant 30 minutes.
La dose recommandée de KEYTRUDA en monothérapie chez les patients pédiatriques âgés de 3 ans et plus atteints d'un LHc ou chez les patients âgés de 12 ans et plus atteints d'un mélanome est de 2 mg/kg de poids corporel (jusqu'à un maximum de 200 mg) toutes les 3 semaines, administrée en perfusion intraveineuse pendant 30 minutes.
Pour une utilisation en association, voir le Résumé des Caractéristiques du Produit (RCP) des traitements concomitants.
Les patients doivent être traités par KEYTRUDA jusqu'à progression de la maladie ou toxicité inacceptable (et jusqu'à la durée maximale du traitement si spécifiée pour une indication). Des réponses atypiques (c'est- à-dire une augmentation initiale et transitoire de la taille de la tumeur ou l'apparition de nouvelles lésions de petite taille durant les premiers mois, suivies d'une régression de la tumeur) ont été observées. Chez les patients cliniquement stables présentant une progression initiale de la maladie, il est recommandé de poursuivre le traitement jusqu'à ce que la progression soit confirmée.
Dans le traitement adjuvant du mélanome ou du carcinome à cellules rénales, KEYTRUDA doit être administré jusqu'à récidive de la maladie, toxicité inacceptable ou pendant une durée allant jusqu'à un an.
Dans le traitement néoadjuvant et adjuvant du CSTN, les patients doivent être traités par KEYTRUDA en néoadjuvant en association à une chimiothérapie à raison de 8 doses de 200 mg toutes les 3 semaines ou 4 doses de 400 mg toutes les 6 semaines ou jusqu'à progression de la maladie empêchant une chirurgie définitive ou toxicité inacceptable, suivi d'un traitement adjuvant par KEYTRUDA en monothérapie à raison de 9 doses de 200 mg toutes les 3 semaines ou 5 doses de 400 mg toutes les 6 semaines ou jusqu'à récidive de la maladie ou toxicité inacceptable. Les patients dont la progression de la maladie empêche une chirurgie définitive ou qui présentent une toxicité inacceptable liée à KEYTRUDA en traitement néoadjuvant en association à une chimiothérapie ne doivent pas recevoir KEYTRUDA en monothérapie en traitement adjuvant.
Suspension ou arrêt définitif du traitement (voir aussi rubrique Mises en garde spéciales et précautions d'emploi)
Aucune réduction de dose de KEYTRUDA n'est recommandée. KEYTRUDA doit être suspendu ou arrêté pour gérer les effets indésirables tels que décrit dans le tableau 1.
Tableau 1 : Modifications de traitement recommandées pour KEYTRUDA
| Effets indésirables d'origine immunologique | Sévérité | Modification de traitement |
| Pneumopathie inflammatoire | Grade 2 | Suspension jusqu'à amélioration des effets indésirables aux Grades 0-1* |
| Grades 3 ou 4, ou Grade 2 récurrent | Arrêt définitif | |
| Colite | Grades 2 ou 3 | Suspension jusqu'à amélioration des effets indésirables aux Grades 0-1* |
| Grade 4 ou Grade 3 récurrent | Arrêt définitif | |
| Néphrite | Grade 2 avec créatinine > 1,5 à ≤ 3 fois la limite supérieure de la normale (LSN) | Suspension jusqu'àamélioration des effets indésirables aux Grades 0-1* |
| Grade ≥ 3 avec créatinine > 3 fois la LSN | Arrêt définitif | |
| Endocrinopathies | Insuffisance surrénalienne et hypophysite de Grade 2 | Suspension du traitementjusqu'au contrôle par traitementhormonal substitutif |
| Insuffisance surrénalienne ou hypophysite symptomatique de Grades 3 ou 4 Diabète de type I associé à une hyperglycémie de Grade ≥ 3 (glucose > 250 mg/dL ou> 13,9 mmol/L) ou associé à une acidocétose Hyperthyroïdie de Grade ≥ 3 | Suspension jusqu'à amélioration des effets indésirables aux Grades 0-1* Pour les patients présentant des endocrinopathies de Grade 3 ou Grade 4 qui se sont amélioréesjusqu'au Grade 2 ou inférieur etsont contrôlées par traitement hormonal substitutif, si indiqué, la poursuite de pembrolizumab peut être envisagée si nécessaire, après diminution progressive de la corticothérapie. Sinon, letraitement doit être arrêté |
| Effets indésirables d'origine immunologique | Sévérité | Modification de traitement |
| définitivement. | ||
| Hypothyroïdie | L'hypothyroïdie peut être priseen charge par traitement hormonal substitutif sans interruption du traitement. | |
| Hépatite NOTE : pour les patients atteints d'un CCR traités par pembrolizumab en association à l'axitinib présentant des augmentations des enzymes hépatiques, voir les recommandations de posologie à la suite de cetableau. | Grade 2 avec aspartate aminotransférase (ASAT) ou alanine aminotransférase (ALAT) > 3 à 5 fois la LSN ou bilirubine totale > 1,5 à 3 fois la LSN | Suspension jusqu'à amélioration des effets indésirables aux Grades 0-1* |
| Grade ≥ 3 avec ASAT ou ALAT > 5 foisla LSN ou bilirubine totale > 3 fois la LSN | Arrêt définitif | |
| En cas de métastases hépatiques avec une augmentation initiale de Grade 2 des ASAT ou des ALAT, hépatite avec augmentation des ASAT ou des ALAT≥ 50 % pendant ≥ 1 semaine | Arrêt définitif | |
| Réactions cutanées | Grade 3 ou syndrome de Stevens- Johnson (SSJ) ou nécrolyse épidermiquetoxique (NET) suspectés | Suspension jusqu'à amélioration des effets indésirables aux Grades 0-1* |
| Grade 4 ou SSJ ou NET confirmés | Arrêt définitif | |
| Autres effets indésirablesd'origine immunologique | Selon la sévérité et le type de réaction (Grade 2 ou Grade 3) | Suspension jusqu'à amélioration des effets indésirables aux Grades 0-1* |
| Myocardite de Grades 3 ou 4 Encéphalite de Grades 3 ou 4 Syndrome de Guillain-Barré de Grades 3 ou 4 | Arrêt définitif | |
| Grade 4 ou Grade 3 récurrent | Arrêt définitif | |
| Réactions liées à laperfusion | Grades 3 ou 4 | Arrêt définitif |
* Si une toxicité liée au traitement ne s'améliore pas jusqu'aux Grades 0-1 dans les 12 semaines après la dernière administration de KEYTRUDA, ou si la dose de corticostéroïdes ne peut pas être réduite dans les 12 semaines à une dose ≤ 10 mg de prednisone ou équivalent par jour, KEYTRUDA doit être arrêté définitivement.
La sécurité de la ré-administration d'un traitement par pembrolizumab chez les patients ayant précédemment présenté une myocardite d'origine immunologique n'est pas connue.
KEYTRUDA, en monothérapie ou en association, doit être arrêté définitivement en cas d'ef ets indésirables d'origine immunologique de Grade 4 ou de Grade 3 récurrent, sauf indication contraire dans le Tableau 1.
En cas de toxicité hématologique de Grade 4, uniquement chez les patients atteints d'un LHc, KEYTRUDA doit être suspendu jusqu'à amélioration des ef ets indésirables aux Grades 0-1.
KEYTRUDA en association à l'axitinib dans le CCR
Chez les patients atteints d'un CCR traités par
KEYTRUDA en association à l'axitinib, voir le RCP concernant la
posologie de l'axitinib. En association à pembrolizumab, l'augmentation
de dose d'axitinib au-delà de la dose initiale de 5 mg peut être envisagée à intervalles de six semaines ou plus (voir rubrique Propriétés pharmacodynamiques).
En cas d'augmentation des enzymes hépatiques chez les patients atteints d'un CCR traités par KEYTRUDA en association à l'axitinib :
- Si les ALAT ou les ASAT sont ≥ 3 fois la LSN mais < 10 fois la LSN sans bilirubine totale concomitante ≥ 2 fois la LSN, KEYTRUDA et l'axitinib doivent être suspendus jusqu'à amélioration de ces ef ets indésirables jusqu'aux Grades 0-1. Une corticothérapie peut être envisagée. La réintroduction d'un seul médicament ou la réintroduction séquentielle des deux médicaments après amélioration peut être envisagée. En cas de réintroduction de l'axitinib, une réduction de la dose peut être envisagée comme mentionné dans le RCP de l'axitinib.
- Si les ALAT ou les ASAT sont ≥ 10 fois la LSN ou > 3 fois la LSN avec une bilirubine totale concomitante ≥ 2 fois la LSN, KEYTRUDA et l'axitinib doivent être arrêtés définitivement et une corticothérapie peut être envisagée.
KEYTRUDA en association au lenvatinib
Lorsqu'il est utilisé en association au lenvatinib, l'un ou les deux médicaments doivent être interrompus selon le cas. Lenvatinib doit être suspendu, sa dose doit être réduite ou il doit être arrêté conformément aux instructions concernant l'association au pembrolizumab dans le RCP du lenvatinib . Aucune réduction de dose n'est recommandée pour KEYTRUDA.
Les patients traités par KEYTRUDA doivent avoir reçu la carte de signalement patient et avoir été informés des risques de KEYTRUDA (voir également la notice).
Populations particulières
Personnes âgées
Aucune adaptation posologique n'est nécessaire chez les patients âgés de ≥ 65 ans (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacodynamiques).
Insuffisance rénale
Aucune adaptation posologique n'est nécessaire pour les patients présentant une insuf isance rénale légère ou modérée. KEYTRUDA n'a pas été étudié chez les patients présentant une insuf isance rénale sévère (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacocinétiques).
Insuffisance hépatique
Aucune adaptation posologique n'est nécessaire pour les patients présentant une insuf isance hépatique légère ou modérée. KEYTRUDA n'a pas été étudié chez les patients présentant une insuf isance hépatique sévère (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l'ef icacité de KEYTRUDA chez les enfants de moins de 18 ans n'ont pas été établies, sauf pour les patients pédiatriques atteints d'un mélanome ou d'un LHc. Les données actuellement disponibles sont décrites en rubriques Effets indésirables, Propriétés pharmacodynamiques et Propriétés pharmacocinétiques.
Mode d'administration
KEYTRUDA est à usage intraveineux. Il doit être administré par perfusion sur une durée de 30 minutes. KEYTRUDA ne doit pas être administré en injection rapide ou en bolus.
Lorsque KEYTRUDA est utilisé en association à une chimiothérapie intraveineuse, KEYTRUDA doit être administré en premier.
Pour les instructions concernant la dilution du médicament avant administration, voir la rubrique Précautions particulières d'élimination et de manipulation.
Durée de conservation :
Flacon non ouvert
2 ans.
Après préparation de la perfusion
D'un point de vue microbiologique, le produit, une fois dilué, doit être utilisé immédiatement. La solution diluée ne doit pas être congelée. Si elle n'est pas utilisée immédiatement, la stabilité chimique et physique de KEYTRUDA en cours d'utilisation a été démontrée pendant 96 heures entre 2°C et 8°C. Cette durée de 96 heures peut inclure jusqu'à 6 heures à température ambiante (à 25°C ou en dessous). En cas de réfrigération, il faut laisser les flacons et/ou les poches de perfusion intraveineuse revenir à température ambiante avant utilisation.
Précautions particulières de conservation :
A conserver au réfrigérateur (2°C - 8°C).
Ne pas congeler.
A conserver dans la boîte d'origine, à l'abri de la lumière.
Pour les conditions de conservation après dilution du médicament, voir la rubrique Durée de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments excepté ceux mentionnés en rubrique Précautions particulières d'élimination et de manipulation.
Il n'existe aucune information sur le surdosage avec pembrolizumab.
En cas de surdosage, les patients doivent être étroitement surveillés pour rechercher des signes ou symptômes évocateurs d'effets indésirables et un traitement symptomatique approprié doit être instauré.
Classe pharmacothérapeutique : Agents antinéoplasiques, inhibiteurs de PD-1/PDL-1 (Programmed cell death protein 1/death ligand 1). Code ATC: L01FF02
Mécanisme d'action
KEYTRUDA est un anticorps monoclonal humanisé qui se lie au récepteur PD-1 (programmed cell death-1) et bloque son interaction avec les ligands PD-L1 et PD-L2. Le récepteur PD-1 est un régulateur négatif de l'activité des cellules T, qui a montré son implication dans le contrôle des réponses immunitaires des cellules T. KEYTRUDA potentialise les réponses des cellules T, y compris les réponses anti-tumorales, par le blocage de la liaison de PD-1 avec PD-L1 et PD-L2, qui sont exprimés dans les cellules présentatrices d'antigène et peuvent être exprimés par les tumeurs ou par d'autres cellules du microenvironnement tumoral.
L'effet anti-angiogénique du lenvatinib (multi ITK) en association avec l'effet de stimulation immunitaire du pembrolizumab (anti PD-1) entraîne un microenvironnement tumoral avec une plus grande activation des cellules T pour aider à contrer la résistance primaire et acquise à l'immunothérapie et peut améliorer les réponses tumorales par rapport à l'un des traitements seuls. Dans les modèles précliniques murins, les inhibiteurs de PD-1 plus ITK ont démontré une activité anti-tumorale améliorée par rapport à l'un des agents seuls.
Efficacité et sécurité clinique
Des doses de pembrolizumab de 2 mg/kg de poids corporel toutes les 3 semaines, 10 mg/kg de poids corporel toutes les 3 semaines et 10 mg/kg de poids corporel toutes les 2 semaines ont été évaluées dans des études cliniques effectuées dans le mélanome ou dans le CBNPC préalablement traité. Sur la base de la modélisation et de la simulation des relations dose/exposition pour l'ef icacité et la tolérance pour le pembrolizumab, il n'y a pas de dif érences cliniquement significatives d'ef icacité ou de tolérance entre les doses de 200 mg toutes les 3 semaines, de 2 mg/kg de poids corporel toutes les 3 semaines et de 400 mg toutes les 6 semaines (voir rubrique Posologie et mode d'administration).
Mélanome
KEYNOTE-006 : Etude contrôlée chez des patients atteints d'un mélanome naïfs de traitement par ipilimumab
L'efficacité et la tolérance de pembrolizumab ont été étudiées dans KEYNOTE-006, une étude de phase III multicentrique, en ouvert, contrôlée, dans le traitement du mélanome avancé chez des patients naïfs d'ipilimumab. Les patients ont été randomisés (1:1:1) pour recevoir pembrolizumab à une dose de 10 mg/kg de poids corporel toutes les 2 (n = 279) ou 3 semaines (n = 277) ou ipilimumab 3 mg/kg de poids corporel toutes les 3 semaines (n = 278). Pour les patients atteints d'un mélanome présentant une mutation BRAF V600E, un traitement préalable par un inhibiteur de BRAF n'était pas requis.
Les patients étaient traités par pembrolizumab jusqu'à progression de la maladie ou toxicité inacceptable. Les patients cliniquement stables avec une preuve initiale de progression de la maladie pouvaient rester sous traitement jusqu'à ce que la progression de la maladie soit confirmée. Une évaluation de la réponse tumorale était réalisée à 12 semaines, puis toutes les 6 semaines jusqu'à la Semaine 48 et ensuite toutes les 12 semaines.
Sur les 834 patients, 60 % étaient des hommes, 44 % étaient âgés ≥ 65 ans (l'âge médian était de 62 ans [18 - 89 ans]) et 98 % étaient de type caucasien. Soixante-cinq pourcent des patients présentaient un stade M1c, 9 % avaient un antécédent de métastases cérébrales, 66 % n'avaient reçu aucun traitement antérieur et 34 % avaient reçu un traitement antérieur. Trente-et-un pourcent avaient un statut de performance ECOG de 1, 69 % avaient un statut de performance ECOG de 0, et 32 % avaient des LDH élevées. Des mutations BRAF étaient rapportées chez 302 (36 %) patients. Parmi les patients porteurs de tumeurs avec mutation BRAF, 139 (46 %) avaient reçu un traitement préalable par inhibiteur de BRAF.
Les critères principaux d'évaluation de l'ef icacité étaient la survie sans progression (Progression Free Survival PFS, évaluée selon une revue clinico-radiologique (Integrated Radiology and Oncology Assessment [IRO] en utilisant les critères RECIST [Response Evaluation Criteria in Solid Tumors], version 1.1) et lasurvie globale (Overall Survival OS). Les critères secondaires d'évaluation de l'e f icacité étaient le taux de réponse objective (Objective Response Rate ORR) et la durée de réponse. Le tableau 3 résume les critères clés d'ef icacité chez les patients naïfs de traitement par ipilimumab lors de l'analyse finale ef ectuée après un suivi minimum de 21 mois. Les courbes de Kaplan-Meier pour l'OS et la PFS, basées sur l'analyse finale, sont représentées en Figures 1 et 2.
Tableau 3 : Résultats d'efficacité dans l'étude KEYNOTE-006
| Critère d'évaluation | Pembrolizumab 10 mg/kg de poids corporel toutes les 3 semainesn = 277 | Pembrolizumab 10 mg/kg de poids corporel toutes les 2 semainesn = 279 | Ipilimumab 3 mg/kg depoids corporel toutes les3 semainesn = 278 |
| OS | |||
| Nombre de patients avec évènement (%) | 119 (43 %) | 122 (44 %) | 142 (51 %) |
| Hazard ratio* (IC 95 %) | 0,68 (0,53 - 0,86) | 0,68 (0,53 - 0,87) | --- |
| Valeur de p† | < 0,001 | < 0,001 | --- |
| Médiane en mois (IC 95%) | Non atteinte(24 - ND) | Non atteinte(22 - ND) | 16(14 - 22) |
| PFS | |||
| Nombre de patients avec évènement (%) | 183 (66 %) | 181 (65 %) | 202 (73 %) |
| Hazard ratio* (IC 95%) | 0,61 (0,50 - 0,75) | 0,61 (0,50 - 0,75) | --- |
| Valeur de p† | < 0,001 | < 0,001 | --- |
| Médiane en mois (IC 95%) | 4,1(2,9 - 7,2) | 5,6(3,4 - 8,2) | 2,8(2,8 - 2,9) |
| Meilleure réponse objective | |||
| ORR % (IC 95 %) | 36 %(30 - 42) | 37 %(31 - 43) | 13 %(10 - 18) |
| Réponse complète | 13 % | 12 % | 5 % |
| Réponse partielle | 23 % | 25 % | 8 % |
| Durée de réponse‡ | |||
| Médiane en mois (intervalle) | Non atteinte (2,0 - 22,8+) | Non atteinte (1,8 - 22,8+) | Non atteinte (1,1+ - 23,8+) |
| % en cours à 18 mois | 68 %§ | 71 %§ | 70 %§ |
* Hazard ratio (pembrolizumab
comparé à ipilimumab) sur la base du modèle « Cox proportional hazard »
stratifié
† Sur la base du test de log rank stratifié
‡ Sur la base des patients ayant une meilleure réponse objective telle que
réponse complète ou partielle confirmées
§ Sur la base de l'estimation de Kaplan Meier
ND = Non disponible
Figure 1 : Courbe de Kaplan-Meier pour la
survie globale par bras de traitement dans
l'étude KEYNOTE-006 (population en intention de traiter)
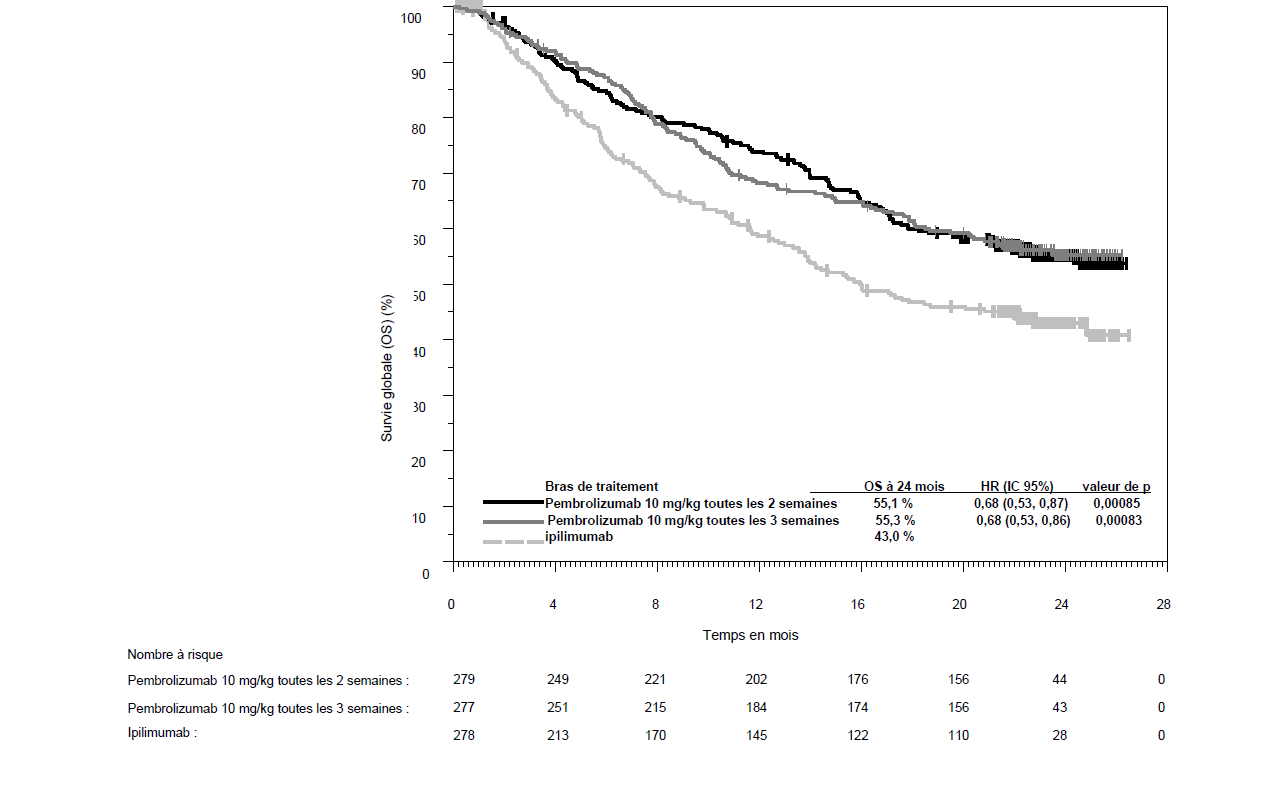
Figure 2 : Courbe de Kaplan-Meier pour la
survie sans progression par bras de traitement dans
l'étude KEYNOTE-006 (population en intention de
traiter)
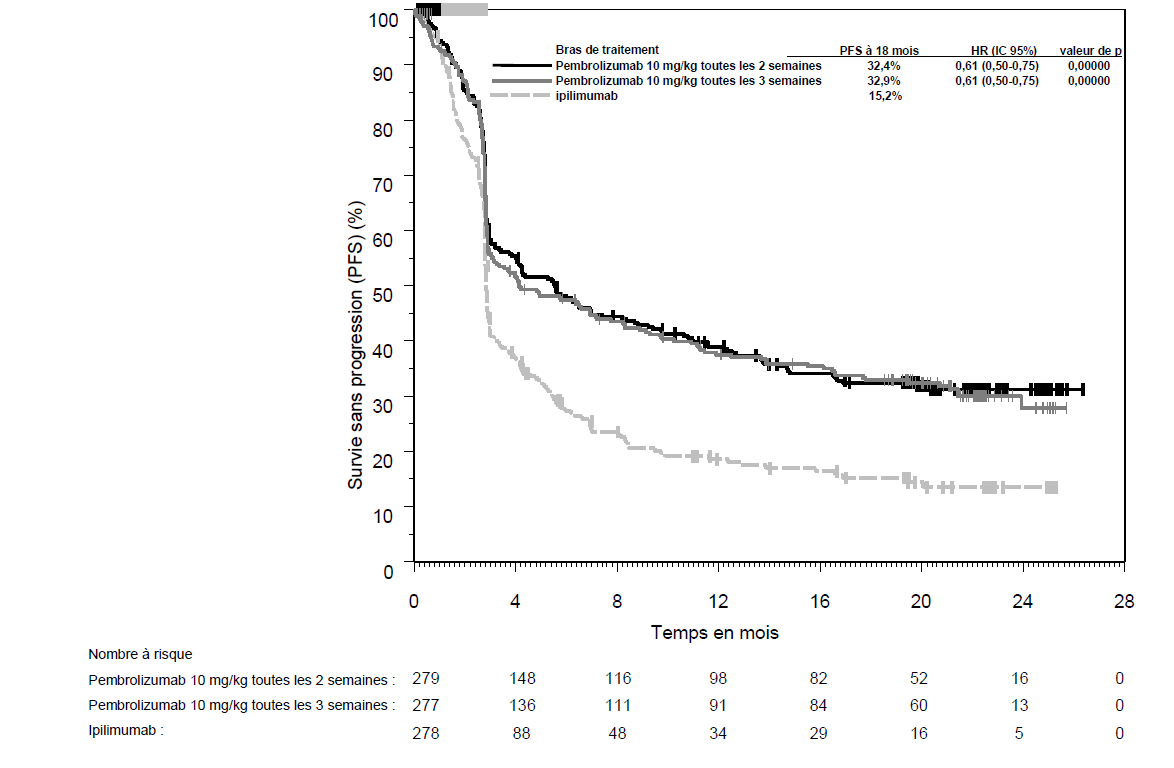
KEYNOTE-002 : Etude contrôlée chez des patients atteints d'un mélanome précédemment traités par ipilimumab
L'efficacité et la tolérance de pembrolizumab ont été étudiées dans KEYNOTE-002, une étude multicentrique, en double aveugle, contrôlée dans le traitement du mélanome avancé chez des patients précédemment traités par ipilimumab et, en cas de mutation BRAF V600, par un inhibiteur de BRAF ou de MEK. Les patients ont été randomisés (1:1:1) pour recevoir pembrolizumab à une dose de 2 (n = 180) ou 10 mg/kg de poids corporel (n = 181) toutes les 3 semaines ou une chimiothérapie (n = 179, incluant dacarbazine, témozolomide, carboplatine, paclitaxel, ou carboplatine + paclitaxel). L'étude excluait les patients atteints d'une maladie auto-immune ou ceux recevant un immunosuppresseur ; d'autres critères d'exclusion étaient : antécédent d'ef et indésirable d'origine immunologique sévère ou engageant le pronostic vital sous ipilimumab, défini comme toute toxicité de Grade 4 ou une toxicité de Grade 3 nécessitant une traitement par corticostéroïde (> 10 mg/jour de prednisone ou une dose équivalente) pendant plus de 12 semaines ; un effet indésirable en cours de Grade ≥ 2 d'un traitement préalable par ipilimumab ; une hypersensibilité sévère antérieure avec d'autres anticorps monoclonaux ; un antécédent de pneumopathie inflammatoire ou de pneumopathie interstitielle ; une infection par le VIH, le virus de l'hépatite B ou de l'hépatite C ; un statut de performance ECOG ≥ 2.
Les patients étaient traités par pembrolizumab jusqu'à progression de la maladie ou toxicité inacceptable. Les patients cliniquement stables avec une preuve initiale de progression de la maladie pouvaient rester sous traitement jusqu'à ce que la progression de la maladie soit confirmée. Une évaluation de la réponse tumorale était réalisée à 12 semaines, puis toutes les 6 semaines jusqu'à la Semaine 48 et ensuite toutes les 12 semaines. Les patients recevant la chimiothérapie et ayant une progression de leur maladie vérifiée de façon indépendante après la première évaluation programmée de la maladie pouvaient bénéficier d'un cross-over et recevoir 2 mg/kg de poids corporel ou 10 mg/kg de poids corporel de pembrolizumab toutes les 3 semaines, en double aveugle.
Sur les 540 patients, 61 % étaient des hommes, 43 % avaient ≥ 65 ans (l'âge médian était de 62 ans [de 15 à 89 ans]) et 98 % étaient de type caucasien. Quatre-vingt-deux pourcent présentaient un stade M1c, 73 % avaient précédemment reçu au moins 2 lignes de traitement systémique du mélanome avancé et 32 % en avaient reçu 3 ou plus. Quarante-cinq pourcent avaient un statut de performance ECOG de 1, 40 % avaient des LDH élevés et 23 % avaient une tumeur mutée BRAF.
Les critères principaux d'évaluation de l'efficacité étaient la PFS, évaluée selon une revue clinico- radiologique (IRO) en utilisant les critères RECIST version 1.1, et l'OS. Les critères secondaires d'évaluation de l'ef icacité étaient l'ORR et la durée de la réponse. Le tableau 4 résume les critères clés d'efficacité lors de l'analyse finale chez les patients précédemment traités par ipilimumab, et la courbe de Kaplan-Meier de PFS est représentée en Figure 3. Les deux bras pembrolizumab étaient supérieurs à la chimiothérapie en termes de PFS, et il n'y avait pas de différence entre les doses de pembrolizumab. Lors de l'analyse finale de l'OS, non ajustée pour les ef ets confondants potentiels du cross-over, il n'y avait pas de différence statistiquement significative entre pembrolizumab et la chimiothérapie. Parmi les patients randomisés dans le bras chimiothérapie, 55 % ont bénéficié d'un cross-over et ont donc reçu un traitement par pembrolizumab.
Tableau 4 : Résultats d'efficacité dans l'étude KEYNOTE-002
| Critère d'évaluation | Pembrolizumab 2 mg/kg de poids corporel toutes les 3 semainesn = 180 | Pembrolizumab 10 mg/kg de poids corporel toutes les3 semainesn = 181 | Chimiothérapien = 179 | |
| PFS | ||||
| Nombre de patients avec évènement (%) | 150 (83 %) | 144 (80 %) | 172 (96 %) | |
| Hazard ratio* (IC 95 %) | 0,58 (0,46 - 0,73) | 0,47 (0,37 - 0,60) | --- | |
| Valeur de p† | < 0,001 | < 0,001 | --- | |
| Médiane en mois (IC 95 %) | 2,9 (2,8 - 3,8) | 3,0 (2,8 - 5,2) | 2,8 (2,6 - 2,8) | |
| OS | ||||
| Nombre de patients avec évènement (%) | 123 (68 %) | 117 (65 %) | 128 (72 %) | |
| Hazard ratio* (IC 95 %) | 0,86 (0,67 - 1,10) | 0,74 (0,57 - 0,96) | --- | |
| Valeur de p† | 0,1173 | 0,0106‡ | --- | |
| Médiane en mois (IC 95 %) | 13,4 (11,0 - 16,4) | 14,7 (11,3 - 19,5) | 11,0 (8,9 - 13,8) | |
| Meilleure réponse objective | ||||
| ORR % (IC 95 %) | 22 % (16 - 29) | 28 % (21 - 35) | 5 % (2 - 9) | |
| Réponse complète | 3 % | 7 % | 0 % | |
| Réponse partielle | 19 % | 20 % | 5 % | |
| Durée de réponse§ | ||||
| Médiane en mois (intervalle) | 22,8(1,4+ - 25,3+) | Non atteinte (1,1+ - 28,3+) | 6,8(2,8 - 11.3) | |
| % en cours à 12 mois | 73 %¶ | 79 %¶ | 0 %¶ | |
* Hazard ratio (pembrolizumab
comparé à la chimiothérapie) sur la base du modèle « Cox proportional hazard »
stratifié
† Sur la base du test de log rank stratifié
‡ Non statistiquement significatif après ajustement pour multiplicité
§ Sur la base des patients ayant une meilleure réponse objective telle que
réponse complète ou partielle confirmée, issue de l'analyse finale
¶ Sur la base de l'estimation de Kaplan-Meier
Figure 3 : Courbe de Kaplan-Meier pour la
survie sans progression par bras de traitement de l'étude
KEYNOTE-002 (population en intention de traiter)
KEYNOTE-001 : Etude ouverte chez les patients atteints d'un mélanome, naïfs de traitement et précédemment traités par ipilimumab
L'efficacité et la tolérance de pembrolizumab chez les patients atteints de mélanome avancé ont été étudiées dans une étude ouverte non contrôlée, KEYNOTE-001. L'efficacité a été évaluée pour 276 patients de deux cohortes définies, l'une ayant inclus des patients précédemment traités par ipilimumab (et par un inhibiteur de BRAF ou de MEK en cas de mutation BRAF V600) et l'autre ayant inclus des patients naïfs de traitementpar ipilimumab. Les patients ont été répartis aléatoirement pour recevoir pembrolizumab à la dose de 2 mg/kg de poids corporel toutes les 3 semaines ou 10 mg/kg de poids corporel toutes les 3 semaines. Les patients ont été traités par pembrolizumab jusqu'à progression de la maladie ou toxicité inacceptable. Les patients cliniquement stables avec une preuve initiale de progression de la maladie pouvaient rester sous traitement jusqu'à ce que la progression de la maladie soit confirmée. Les critères d'exclusion étaient similaires à ceux de KEYNOTE-002.
Sur les 89 patients ayant reçu 2 mg/kg de poids corporel de pembrolizumab qui étaient précédemment traités par ipilimumab, 53 % étaient des hommes, 33 % étaient âgés de ≥ 65 ans et l'âge médian était de 59 ans (de 18 à 88 ans). Tous les patients étaient de type caucasien, sauf deux. Quatre-vingt-quatre pourcent présentaient un stade M1c et 8 % des patients avaient des antécédents de métastases cérébrales. Soixante-dix pourcent avaient eu au moins 2 traitements systémiques antérieurs pour leur mélanome avancé et 35 % des patients en avaient eu 3 ou plus. Des mutations BRAF ont été rapportées pour 13 % de la population del'étude. Tous les patients présentant des tumeurs mutées BRAF avaient été préalablement traités par un inhibiteur de BRAF.
Sur les 51 patients recevant 2 mg/kg de poids corporel de pembrolizumab qui étaient naïfs de traitement par ipilimumab, 63 % étaient des hommes, 35 % étaient âgés de ≥ 65 ans et l'âge médian était de 60 ans (de 35 à 80 ans). Tous les patients étaient de type caucasien, sauf un. Soixante-trois pourcent présentaient un stade M1c et 2 % des patients avaient des antécédents de métastases cérébrales. Quarante-cinq pourcent n'avaient reçu aucun traitement antérieur pour le mélanome avancé. Des mutations BRAF ont été rapportées chez 20 (39 %) patients. Parmi les patients présentant des tumeurs mutées BRAF, 10 (50 %) avaient été préalablement traités par un inhibiteur de BRAF.
Le critère principal d'évaluation de l'ef icacité était l'ORR, tel qu'évalué par une revue indépendante en utilisant RECIST 1.1. Les critères secondaires d'évaluation de l'efficacité étaient le taux de contrôle de la maladie (incluant réponse complète, réponse partielle et maladie stable), la durée de la réponse, la PFS et l'OS. La réponse tumorale a été évaluée à des intervalles de 12 semaines. Le tableau 5 résume les critères clés d'évaluation de l'efficacité chez les patients déjà traités ou naïfs de traitement par ipilimumab, recevant pembrolizumab à une dose de 2 mg/kg de poids corporel, sur la base d'un suivi minimum d'une durée de 30 mois pour tous les patients.
Tableau 5 : Résultats d'ef icacité dans l'étude KEYNOTE-001
| Critère d'évaluation | Pembrolizumab 2 mg/kg de poids corporel toutes les 3 semaines chez les patients déjà traités par ipilimumab n = 89 | Pembrolizumab 2 mg/kg de poids corporel toutes les3 semaines chez les patients naïfs de traitement par ipilimumabn = 51 |
| Meilleure réponse objective* selonIRO† | ||
| Taux de réponse objective (ORR) %,(IC 95 %) | 26 % (17 - 36) | 35 % (22 - 50) |
| Réponse complète | 7 % | 12 % |
| Réponse partielle | 19 % | 24 % |
| Taux de contrôle de la maladie %‡ | 48 % | 49 % |
| Durée de réponse§ | ||
| Médiane en mois (intervalle) | 30,5 (2,8+ - 30,6+) | 27,4 (1,6+ - 31,8+) |
| % en cours à 24 mois¶ | 75 % | 71 % |
| Survie sans progression (PFS) | ||
| Médiane en mois (IC 95 %) | 4,9 (2,8 - 8,3) | 4,7 (2,8 -13,8) |
| PFS à 12 mois | 34 % | 38 % |
| Survie globale (OS) | ||
| Médiane en mois (IC 95 %) | 18,9 (11 - non disponible) | 28,0 (14 - non disponible) |
| OS à 24 mois | 44 % | 56 % |
* Inclut les patients sans maladie
mesurable par radiologie indépendante à l'initiation
† IRO = évaluation intégrée radiologique et oncologique (Independent radiology
and oncologist) utilisant RECIST 1.1
‡ Sur la base de la meilleure réponse de la maladie, stable ou meilleur
§ Sur la base des patients présentant une réponse confirmée par une revue
indépendante, à partir de la date où la réponse a été enregistrée pour la 1ère
fois ; n = 23 pour les patients préalablement traités par ipilimumab ; n = 18
pour les patients naïfs de traitement par ipilimumab
¶ Sur la base de l'estimation de Kaplan-Meier
Les résultats pour les patients précédemment traités par ipilimumab (n = 84) et pour les patients naïfs de traitement par ipilimumab (n = 52) ayant reçu 10 mg/kg de poids corporel de pembrolizumab toutes les 3 semaines étaient similaires à ceux observés chez les patients qui ont reçu 2 mg/kg de poids corporel de pembrolizumab toutes les 3 semaines.
Analyse des sous-populations
Statut de mutation BRAF dans le mélanome
Une analyse en sous-groupes a été réalisée dans le cadre de l'analyse finale de l'étude KEYNOTE-002 chez les patients qui étaient BRAF de type sauvage (n = 414 ; 77 %) ou mutés BRAF avec un traitement BRAF préalable (n = 126 ; 23 %), comme résumée dans le tableau 6.
Tableau 6 : Résultats d'ef icacité selon le statut de mutation BRAF dans l'étude KEYNOTE-002
| BRAF de type sauvage | Mutation BRAF avectraitement BRAF préalable | |||
| Critère d'évaluation | Pembrolizumab 2 mg/kg de poids corporel toutes les3 semaines(n = 136) | Chimiothérapie (n = 137) | Pembrolizumab 2 mg/kg de poids corporel toutes les3 semaines(n = 44) | Chimiothérapie (n = 42) |
| Hazard ratio* de la PFS (IC 95 %) | 0,50 (0,39 - 0,66) | --- | 0,79 (0,50 - 1,25) | --- |
| Hazard ratio*de l'OS(IC 95 %) | 0,78 (0,58 - 1,04) | --- | 1,07 (0,64 - 1,78) | --- |
| ORR % | 26 % | 6 % | 9 % | 0 % |
* Hazard ratio (pembrolizumab comparé à la chimiothérapie) sur la base du modèle « Cox proportional hazard » stratifié
Une analyse en sous-groupes a été réalisée dans le cadre de l'analyse finale de l'étude KEYNOTE-006 chez les patients qui étaient BRAF de type sauvage (n = 525 ; 63 %), mutés BRAF sans traitement BRAF préalable (n = 163 ; 20 %) et mutés BRAF avec traitement BRAF préalable (n = 139 ; 17 %), comme résumée dans le tableau 7.
Tableau 7 : Résultats d'ef icacité selon le statut de mutation BRAF dans l'étude KEYNOTE-006
| BRAF de type sauvage | Mutation BRAF sanstraitement BRAF préalable | Mutation BRAF avectraitement BRAF préalable | ||||
| Critèred'évaluation | Pembrolizumab 10 mg/kg de poids corporel toutesles 2 ou 3 semaines(poolé) | Ipilimumab (n = 170) | Pembrolizumab 10 mg/kg de poids corporel toutesles 2 ou 3 semaines(poolé) | Ipilimumab (n = 55) | Pembrolizumab 10 mg/kg de poids corporel toutesles 2 ou 3 semaines(poolé) | Ipilimumab (n = 52) |
| Hazard ratio* de la PFS (IC 95 %) | 0,61 (0,49 - 0,76) | --- | 0,52 (0,35 - 0,78) | --- | 0,76 (0,51 - 1,14) | --- |
| Hazard ratio*de l'OS(IC 95 %) | 0,68 (0,52 - 0,88) | --- | 0,70 (0,40 - 1,22) | --- | 0,66 (0,41 - 1,04) | --- |
| ORR % | 38 % | 14 % | 41 % | 15 % | 24 % | 10 % |
*Hazard ratio (pembrolizumab comparé à ipilimumab) sur la base du modèle « Cox proportional hazard » stratifié
Statut PD-L1 dans le mélanome
Une analyse en sous-groupes a été réalisée dans le cadre de l'analyse finale de l'étude KEYNOTE-002 chez les patients PD-L1 positifs (expression de PD-L1 sur ≥ 1 % des cellules tumorales et des cellules immunitaires associées à la tumeur par rapport à toutes les cellules tumorales viables - MEL score) vs PD- L1 négatifs. L'expression PD-L1 était testée rétrospectivement par un test d'immunohistochimie (IHC) avec l'anticorps anti-PDL1 22C3. Parmi les patients qui étaient évaluables pour l'expression PD-L1 (79 %), 69 % (n = 294) étaient PD-L1 positifs et 31 % (n = 134) étaient PD-L1 négatifs. Le tableau 8 résume les résultats d'efficacité selon l'expression PD-L1.
Tableau 8 : Résultats d'ef icacité selon l'expression de PD-L1 dans l'étude KEYNOTE-002
| Critèred'évaluation | Pembrolizumab2 mg/kg de poids corporel toutes les 3 semaines | Chimiothérapie | Pembrolizumab2 mg/kg de poids corporel toutes les 3 semaines | Chimiothérapie |
| PD-L1 positif | PD-L1 négatif | |||
| Hazard ratio*de la PFS (IC 95%) | 0,55 (0,40 - 0,76) | --- | 0,81 (0,50 - 1,31) | --- |
| Hazard ratio*de l'OS(IC 95%) | 0,90 (0,63 - 1,28) | --- | 1,18 (0,70 - 1,99) | --- |
| ORR % | 25 % | 4 % | 10 % | 8 % |
*Hazard ratio (pembrolizumab comparé à la chimiothérapie) sur la base du modèle « Cox proportional hazard » stratifié
Une analyse en sous-groupes a été réalisée dans le cadre de l'analyse finale de l'étude KEYNOTE-006 chez les patients PD-L1 positifs (n = 671 ; 80 %) vs PD-L1 négatifs (n = 150 ; 18 %). Parmi les patients qui étaient évaluables pour l'expression PD-L1 (98 %), 82 % étaient PD-L1 positifs et 18 % étaient PD-L1 négatifs. Le tableau 9 résume les résultats d'ef icacité selon l'expression de PD-L1.
Tableau 9 : Résultats d'ef icacité selon l'expression de PD-L1 dans l'étude KEYNOTE-006
| Critère d'évaluation | Pembrolizumab10 mg/kg de poids corporel toutes les 2 ou3 semaines (poolé) | Ipilimumab | Pembrolizumab10 mg/kg de poids corporel toutesles 2 ou3 semaines (poolé) | Ipilimumab |
| PD-L1 positif | PD-L1 négatif | |||
| Hazard ratio* de la PFS(IC 95%) | 0,53 (0,44 - 0,65) | --- | 0,87 (0,58 - 1,30) | --- |
| Hazard ratio*de l'OS(IC 95%) | 0,63 (0,50 - 0,80) | --- | 0,76 (0,48 - 1,19) | --- |
| ORR % | 40 % | 14 % | 24 % | 13 % |
*Hazard ratio (pembrolizumab comparé à ipilimumab) sur la base du modèle « Cox proportional hazard » stratifié
Mélanome oculaire
Chez 20 sujets atteints de mélanome oculaire inclus dans l'étude KEYNOTE-001, aucune réponse objective n'a été rapportée ; une maladie stable a été rapportée chez 6 patients.
KEYNOTE-716 : Etude contrôlée versus placebo pour le traitement adjuvant des patients avec un mélanome réséqué de stade IIB ou IIC
L'efficacité du pembrolizumab a été évaluée dans KEYNOTE-716, une étude multicentrique, randomisée, en double aveugle, contrôlée versus placebo chez des patients atteints d'un mélanome réséqué de stade IIB ou IIC. Un total de 976 patients a été randomisé (1:1) pour recevoir pembrolizumab 200 mg toutes les trois semaines (ou la dose pédiatrique [12 à 17 ans] de 2 mg/kg par voie intraveineuse [jusqu'à un maximum de 200 mg] toutes les trois semaines) (n = 487) ou un placebo (n = 489), sur une durée maximale d'un an ou jusqu'à récidive de la maladie ou toxicité inacceptable. La randomisation a été stratifiée selon le stade T défini par la classification de l'American Joint Committee on Cancer (AJCC), 8ème édition. Les patients présentant une maladie auto-immune active, ou un état clinique nécessitant un traitement immunosuppresseur, ou un mélanome muqueux ou oculaire, étaient inéligibles. Les patients ayant reçu un traitement antérieur pour leur mélanome autre que la chirurgie étaient inéligibles. Les patients ont bénéficié d'une imagerie tous les six mois à compter de la randomisation jusqu'à la 4ème année, et une imagerie à 5 ans à compter de la randomisation ou jusqu'à la récidive, selon l'évènement survenant en premier.
Parmi les 976 patients, les caractéristiques à l'inclusion étaient : âge médian de 61 ans (intervalle : 16 à 87 ; 39 % âgés de 65 ans ou plus ; 2 patients adolescents [un par bras de traitement]) ; 60 % d'hommes ; et statut de performance ECOG de 0 (93 %) et 1 (7 %). Soixante-quatre pour cent avaient une maladie au stade IIB et 35 % au stade IIC.
Le critère principal d'évaluation de l'efficacité était la survie sans récidive (RFS) évaluée par l'investigateur dans la population totale, la RFS étant définie comme le temps entre la date de randomisation et la date de première récidive (locale, régionale ou métastatique à distance) ou de décès, selon ce qui survenait en premier. Les critères secondaires d'évaluation étaient la survie sans métastase à distance (DMFS) et l'OS dans l'ensemble de la population. L'OS n'a pas été formellement évaluée au moment de cette analyse. L'étude a initialement démontré une amélioration statistiquement significative de la RFS (HR 0,65 ; IC 95 % : 0,46 - 0,92 ; p = 0,00658) chez les patients randomisés dans le bras pembrolizumab en comparaison au placebo lors de son analyse intermédiaire prédéfinie. Les résultats de la RFS rapportés lors de son analyse finale pré- définie, réalisée après un suivi médian de 20,5 mois sont résumés dans le tableau 10 et la figure 4. Les résultats de la RFS actualisés après un suivi médian de 26,9 mois étaient cohérents avec l'analyse finale de la RFS pour les patients randomisés dans le bras pembrolizumab en comparaison au placebo (HR 0,64 ; IC 95 % : 0,50 - 0,84). Les résultats de la DMFS lors de l'analyse intermédiaire réalisée après un suivi médian de 26,9 mois sont rapportés dans le tableau 10 et la figure 5.
Tableau 10 : Résultats d'efficacité dans KEYNOTE-716
| Critère d'évaluation | Pembrolizumab 200 mg toutes les 3 semaines n = 487 | Placebon = 489 |
| RFS | ||
| Nombre (%) de patients avec événement | 72 (15 %) | 115 (24 %) |
| Médiane en mois (IC 95%) | NA (NA - NA) | NA (29,9 - NA) |
| Hazard ratio* (IC 95%) | 0,61 (0,45 - 0,82) | |
| Valeur de p (log-rank stratifié) † | 0,00046 | |
| DMFS | ||
| Nombre (%) de patients avec événement | 63 (13 %) | 95 (19 %) |
| Médiane en mois (IC 95%) | NA (NA - NA) | NA (NA - NA) |
| Hazard ratio* (IC 95%) | 0,64 (0,47 - 0,88) | |
| Valeur de p (log-rank stratifié) | 0,00292 | |
* Sur la base
du modèle « Cox proportional hazard » stratifié
† Valeur de p
nominale sur la base du test de log-rank stratifié selon le stade T défini par
la classification de l'American Joint Committee on Cancer (AJCC), 8ème édition
NA = non
atteint
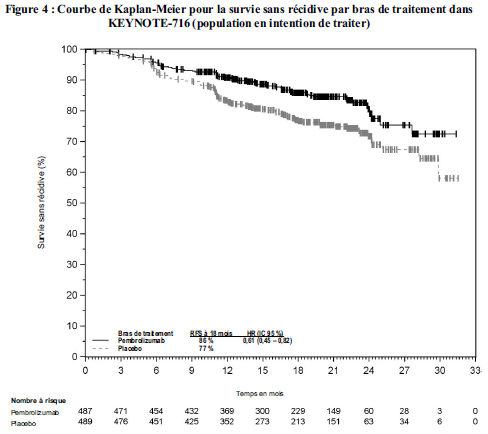
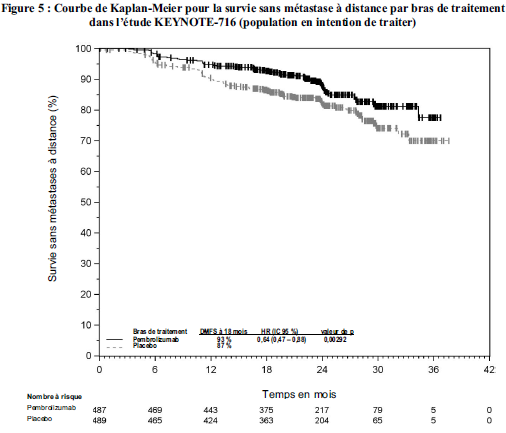
KEYNOTE-054 : Etude contrôlée versus placebo pour le traitement adjuvant des patients avec un mélanome complètement réséqué de stade III
L'efficacité de pembrolizumab a été évaluée dans KEYNOTE-054, une étude multicentrique, randomisée, en double-aveugle, contrôlée versus placebo chez des patients avec un mélanome complètement réséqué de stade IIIA (métastases dans les ganglions lymphatiques > 1 mm), IIIB ou IIIC. Un total de 1 019 patients adultes ont été randomisés (1:1) pour recevoir pembrolizumab 200 mg toutes les trois semaines (n = 514) ou un placebo (n = 505) pendant au maximum un an jusqu'à récidive de la maladie ou toxicité inacceptable. La randomisation était stratifiée, selon le stade de la maladie AJCC, édition 7 (IIIA versus IIIB versus IIIC avec 1-3 ganglions lymphatiques positifs versus IIIC avec ≥ 4 ganglions lymphatiques positifs) et la région géographique (Amérique du Nord, pays européens, Australie et autres pays tels que désignés). Les patients devaient avoir subi un curage ganglionnaire et, si indiqué, une radiothérapie dans les 13 semaines avant le début du traitement. Les patients avec une maladie auto-immune active, ou un état clinique nécessitant un traitement immunosuppresseur, ou avec un mélanome des muqueuses ou oculaire, étaient inéligibles. Les patients ayant déjà reçu un traitement pour un mélanome autre que la chirurgie ou l'interféron pour des mélanomes primitifs épais sans preuve d'atteinte des ganglions lymphatiques étaient inéligibles. Les patients ont bénéficié d'un examen par imagerie toutes les 12 semaines après la première dose de pembrolizumab pendant les deux premières années, puis tous les 6 mois de la 3ème à la 5ème année et, ensuite, annuellement.
Parmi les 1 019 patients, les caractéristiques à l'inclusion étaient : âge médian de 54 ans (25 % âgés de 65 ans ou plus) ; 62 % d'hommes ; et statut de performance ECOG de 0 (94 %) et 1 (6 %). Seize pourcents avaient une maladie au stade IIIA, 46 % au stade IIIB, 18 % au stade IIIC (1-3 ganglions lymphatiques positifs) et 20 % au stade IIIC (≥ 4 ganglions lymphatiques positifs) ; 50 % étaient positifs à la mutation BRAF V600 et 44 % présentaient un gène BRAF de type sauvage. L'expression PD-L1 était testée rétrospectivement par un test d'IHC avec l'anticorps anti-PDL1 22C3 ; 84 % des patients avaient un mélanome PD-L1 positif (expression de PD-L1 dans ≥ 1 % des cellules tumorales et des cellules immunitaires associées à la tumeur par rapport à toutes les cellules tumorales viables). Le même score a été utilisé pour le mélanome métastatique (MEL score).
Les critères principaux d'évaluation de l'efficacité étaient la RFS évaluée par l'investigateur dans la population totale et dans la population avec des tumeurs PD-L1 positives, la RFS étant définie comme le temps entre la date de randomisation et la date de première récidive (locale, régionale ou métastatique à distance) ou de décès, selon ce qui survient en premier. Les critères secondaires d'évaluation étaient la DMFS et l'OS dans l'ensemble de la population et dans la population avec des tumeurs PD-L1 positives. L'OS n'a pas été formellement évaluée au moment de ces analyses. L'étude a initialement démontré une amélioration statistiquement significative de la RFS (HR 0,57 ; IC 98,4 % : 0,43 - 0,74 ; p < 0,0001) chez les patients randomisés dans le bras pembrolizumab en comparaison au placebo lors de son analyse intermédiaire prédéfinie. Les résultats d'ef icacité actualisés avec un suivi médian de 45,5 mois sont résumés dans le tableau 11 et les figures 6 et 7.
Tableau 11 : Résultats d'efficacité dans KEYNOTE-054
| Critère d'évaluation | Pembrolizumab200 mg toutes les 3 semaines n = 514 | Placebon = 505 |
| RFS | ||
| Nombre (%) de patients avec événement | 203 (40 %) | 288 (57 %) |
| Médiane en mois (IC 95%) | NA | 21,4 (16,3 - 27,0) |
| Hazard ratio* (IC 95%) | 0,59 (0,49 - 0,70) | |
| DMFS | ||
| Nombre (%) de patients avecévénement | 173 (34 %) | 245 (49 %) |
| Médiane en mois (IC 95%) | NA | 40,0 (27,7 - NA) |
| Hazard ratio* (IC 95%) | 0,60 (0,49 - 0,73) | |
| Valeur de p (log-rank stratifié) | < 0,0001 | |
* Sur la base du modèle « Cox proportional hazard » stratifié NA = non atteint
Figure 6 : Courbe de Kaplan-Meier pour la survie sans récidive par bras de traitement dans KEYNOTE-054 (population en intention de traiter)
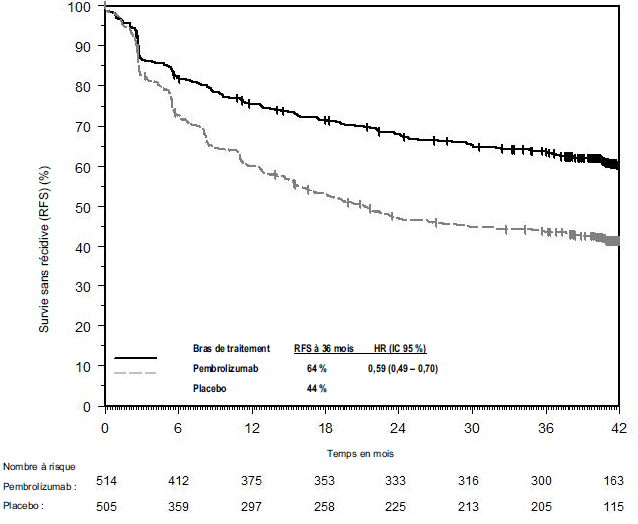
Figure 7 : Courbe de Kaplan-Meier pour la survie sans métastase à distance par bras de traitement dans l'étude KEYNOTE-054 (population en intention de traiter)
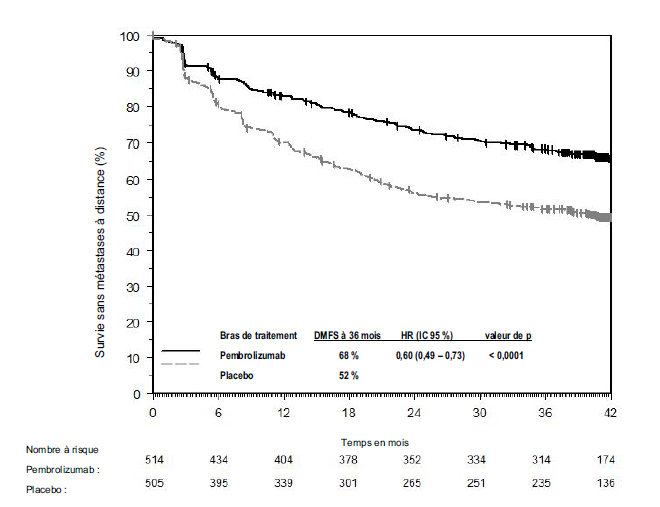
Les bénéfices de la RFS et de la DMFS ont été systématiquement démontrés dans les sous-groupes, y compris selon l'expression de la tumeur PD-L1, le statut mutationnel BRAF et le stade de la maladie (en utilisant l'AJCC édition 7). Ces résultats étaient cohérents lorsqu'ils ont été reclassifiés dans une analyse post hoc selon la classification actuelle de l'AJCC édition 8.
CBNPC
KEYNOTE-024 : Etude contrôlée chez des patients atteints d'un CBNPC naïfs de traitement
L'efficacité et la tolérance de pembrolizumab ont été étudiées dans KEYNOTE-024, une étude multicentrique, en ouvert, contrôlée, dans le traitement des patients atteints d'un CBNPC métastatique non préalablement traité. Les patients devaient exprimer PD-L1 avec un TPS ≥50 % avec le kit PD-L1 IHC 22C3 pharmDxTM. Les patients ont été randomisés (1:1) pour recevoir pembrolizumab à la dose de 200 mg toutes les 3 semaines (n = 154) ou une chimiothérapie à base de sels de platine au choix de l'investigateur (n = 151 incluant pemetrexed + carboplatine, pemetrexed + cisplatine, gemcitabine + cisplatine, gemcitabine + carboplatine ou paclitaxel + carboplatine. Les patients atteints d'un CBNPC non-épidermoïde pouvaient recevoir un traitement de maintenance par pemetrexed). Les patients étaient traités par pembrolizumab jusqu'à apparition d'une toxicité inacceptable ou progression de la maladie. Le traitement pouvait être poursuivi au-delà de la progression de la maladie si le patient était cliniquement stable et si l'investigateur considérait qu'il y avait un bénéfice clinique. Les patients sans progression de leur maladie pouvaient être traités jusqu'à 24 mois. L'étude excluait les patients avec des anomalies génomiques tumorales d'EGFR ou d'ALK ; les patients atteints d'une maladie auto-immune ayant nécessité un traitement systémique dans les 2 ans précédents ; les patients dont l'état clinique nécessitait un traitement immunosuppresseur ; les patients qui avaient reçu plus de 30 Gy d'irradiation thoracique au cours des 26 semaines précédentes. Une évaluation de la réponse tumorale était réalisée toutes les 9 semaines. Les patients sous chimiothérapie qui présentaient une progression de leur maladie vérifiée de manière indépendante pouvaient bénéficier d'un cross-over et recevoir pembrolizumab.
Parmi les 305 patients de l'étude KEYNOTE-024, les caractéristiques à l'inclusion étaient : âge médian de 65 ans (54 % âgés de 65 ans ou plus) ; 61 % d'hommes ; 82 % de type caucasien et 15 % de type asiatique ; le statut de performance ECOG était de 0 et 1 chez 35 % et 65 % des patients, respectivement. Les caractéristiques de la maladie étaient : histologie épidermoïde (18 %) et non-épidermoïde (82 %) ; stade M1 (99 %) ; et métastases cérébrales (9 %).
Le critère principal d'évaluation de l'efficacité était la PFS telle qu'évaluée en aveugle par une revue centralisée indépendante utilisant RECIST 1.1. Les critères secondaires d'évaluation de l'efficacité étaient l'OS et l'ORR (tels qu'évalués en aveugle par une revue centralisée indépendante utilisant RECIST 1.1). Le tableau 12 résume les critères clés de l'efficacité pour l'ensemble de la population en intention de traiter (ITT). Les résultats rapportés pour la PFS et l'ORR sont issus de l'analyse intermédiaire réalisée après un suivi médian de 11 mois. Les résultats rapportés pour l'OS sont issus de l'analyse finale réalisée après un suivi médian de 25 mois.
Tableau 12 : Résultats d'efficacité dans l'étude KEYNOTE-024
| Critère d'évaluation | Pembrolizumab 200 mg toutes les 3 semainesn = 154 | Chimiothérapien = 151 |
| PFS | ||
| Nombre (%) de patients avec évènement | 73 (47 %) | 116 (77 %) |
| Hazard ratio* (IC 95 %) | 0,50 (0,37 ; 0,68) | |
| Valeur de p† | < 0,001 | |
| Médiane en mois (IC 95 %) | 10,3 (6,7 ; ND | 6,0 (4,2 ; 6,2) |
| OS | ||
| Nombre (%) de patients avec évènement | 73 (47 %) | 96 (64 %) |
| Hazard ratio* (IC 95 %) | 0,63 (0,47 ; 0,86) | |
| Valeur de p† | 0,002 | |
| Médiane en mois (IC 95 %) | 30,0 | 14,2 |
| (18,3 ; ND) | (9,8 ; 19,0) | |
| Taux de réponse objective | ||
| ORR % (IC 95 %) | 45 % (37 ; 53) | 28 % (21 ; 36) |
| réponse complète | 4 % | 1 % |
| réponse partielle | 41 % | 27 % |
| Durée de réponse‡ | ||
| Médiane en mois (intervalle) | Non atteinte | 6,3 |
| (1,9+ ; 14,5+) | (2,1+ ; 12,6+) | |
| % avec durée ≥ 6 mois | 88 %§ | 59 %¶ |
*Hazard ratio (pembrolizumab comparé à une chimiothérapie) sur la base du modèle « Cox proportional hazard » stratifié
† Sur la base du test de log rank stratifié
‡ Sur la base des patients avec, pour meilleure réponse objective, une réponse complète ou partielle confirmée
§ Sur la base des estimations de Kaplan-Meier ; inclut 43 patients avec des réponses de 6 mois ou plus
¶ Sur la base des estimations de Kaplan-Meier ; inclut 16 patients avec des réponses de 6 mois ou plus
ND = Non disponible
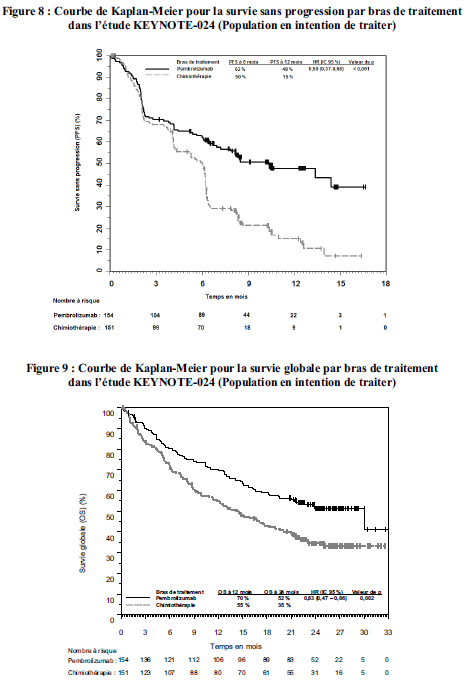
Dans une analyse en sous-groupes, le bénéfice de survie observé avec le pembrolizumab par rapport à la chimiothérapie a été moindre chez le petit nombre de patients n'ayant jamais fumé ; cependant, aucune conclusion définitive ne peut être tirée de ces données en raison du petit nombre de patients.
KEYNOTE-042 : Etude contrôlée chez des patients atteints de CBNPC naïfs de traitement
L'efficacité et la tolérance de pembrolizumab ont également été évaluées dans KEYNOTE-042, une étude multicentrique, contrôlée dans le traitement du CBNPC localement avancé ou métastatique, non préalablement traité. Le design de l'étude était similaire à celui de KEYNOTE-024, sauf que les patients devaient exprimer PD-L1 avec un TPS ≥ 1 % avec le kit PD-L1 IHC 22C3 pharmDxTM. Les patients ont été randomisés (1:1) pour recevoir pembrolizumab à une dose de 200 mg toutes les 3 semaines (n = 637) ou une chimiothérapie à base de sels de platine au choix de l'investigateur (n = 637, incluant pemetrexed+carboplatine ou paclitaxel+carboplatine. Les patients atteints d'un CBNPC non épidermoïde pouvaient recevoir un traitement de maintenance par pemetrexed). Une évaluation de la réponse tumorale était réalisée toutes les 9 semaines pendant les 45 premières semaines et ensuite toutes les 12 semaines.
Parmi les 1 274 patients de l'étude KEYNOTE-042, 599 (47 %) avaient des tumeurs exprimant PD-L1 avec un TPS ≥ 50 % avec le kit PD-L1 IHC 22C3 pharmDxTM. Les caractéristiques à l'inclusion de ces 599 patients comprenaient : âge médian de 63 ans (45 % âgés de 65 ans ou plus) ; 69 % d'hommes ; 63 % de type caucasien et 32 % de type asiatique ; 17 % de type hispanique ou latino-américain ; et le statut de performance ECOG était de 0 et 1 chez 31 % et 69 % des patients, respectivement. Les caractéristiques de la maladie étaient : histologie épidermoïde (37 %) et non-épidermoïde (63 %) ; stade IIIA (0,8 %), stade IIIB (9 %), stade IV (90 %) ; et métastases cérébrales traitées (6 %).
Le critère principal d'évaluation de l'efficacité était l'OS. Les critères secondaires d'évaluation de l'ef icacité étaient la PFS et l'ORR (tels qu'évalués en aveugle par une revue centralisée indépendante utilisant RECIST 1.1). L'étude a démontré une amélioration statistiquement significative de l'OS pour les patients randomisés dans le groupe pembrolizumab en monothérapie dont les tumeurs exprimaient PD-L1 avec un TPS ≥ 1%, par rapport à la chimiothérapie (HR 0,82 ; IC 95 % : 0,71 - 0,93 lors de l'analyse finale) et chez les patients randomisés dans le groupe pembrolizumab en monothérapie dont les tumeurs exprimaient PD-L1 avec un TPS ≥ 50%, par rapport à la chimiothérapie. Le tableau 13 résume les critères clés de l'efficacité pour la population avec un TPS ≥50% lors de l'analyse finale réalisée après un suivi médian de 15,4 mois. La courbe de Kaplan Meier de l'OS pour la population avec un TPS ≥ 50% basée sur l'analyse finale est représentée dans la Figure 10.
Tableau 13 : Résultats d'efficacité (PD-L1 TPS ≥ 50%) dans l'étude KEYNOTE-042
| Critère d'évaluation | Pembrolizumab 200 toutes les3 semaines n = 299 | Chimiothérapie n = 300 |
| OS | ||
| Nombre (%) de patients avec événement | 180 (60 %) | 220 (73 %) |
| Hazard ratio* (IC 95 %) | 0,70 (0,58 - 0,86) | |
| Valeur de p† | 0,0003 | |
| Médiane en mois (IC 95 %) | 20,0 (15,9 - 24,2) | 12,2 (10,4 - 14,6) |
| PFS | ||
| Nombre (%) de patients avec événement | 238 (80 %) | 250 (83 %) |
| Hazard ratio* (IC 95 %) | 0,84 (0,70 - 1,01) | |
| Médiane en mois (IC 95 %) | 6,5 (5,9 - 8,5) | 6,4 (6,2 - 7,2) |
| Taux de réponse objective | ||
| ORR % (IC 95 %) | 39 % (34 - 45) | 32 % (27 - 38) |
| Réponse complète | 1 % | 0.3 % |
| Réponse partielle | 38 % | 32 % |
| Durée de réponse‡ | ||
| Médiane en mois (intervalle) | 22,0(2,1+ - 36,5+) | 10,8(1,8+ - 30,4+) |
| % avec durée ≥ 18 mois | 57 % | 34 % |
* Hazard ratio (pembrolizumab comparé à la chimiothérapie) sur la base du modèle « Cox proportional hazard » stratifié
† Sur la base du test de log rank stratifié
‡ Sur la base des patients avec, pour meilleure réponse objective, une réponse complète ou partielle confirmée
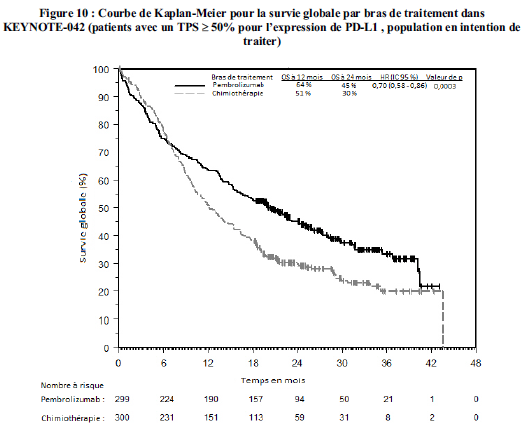
Les résultats d'une analyse post hoc exploratoire en sous-groupe indiquaient une tendance vers un moindre bénéfice de survie avec le pembrolizumab par rapport à la chimiothérapie chez des patients n'ayant jamais fumé, à la fois dans les 4 premiers mois et pendant toute la durée de traitement. Cependant, en raison de la nature exploratoire de cette analyse en sous-groupe, aucune conclusion définitive ne peut être tirée.
KEYNOTE-189 : Etude contrôlée du traitement en association chez des patients naïfs de traitement atteints d'un CBNPC non-épidermoïde
L'efficacité de pembrolizumab en association à une chimiothérapie pemetrexed et sel de platine a été étudiée dans KEYNOTE-189, une étude multicentrique, randomisée, contrôlée par traitement actif, en double-aveugle. Les principaux critères d'éligibilité étaient un CBNPC métastatique non-épidermoïde, l'absence de traitement systémique antérieur pour le CBNPC métastatique et l'absence d'anomalies génomiques tumorales d'EGFR ou d'ALK. Les patients atteints d'une maladie auto-immune ayant nécessité un traitement systémique dans les 2 ans précédents, ceux dont l'état clinique nécessitait un traitement immunosuppresseur ou ceux qui avaient reçu plus de 30 Gy d'irradiation thoracique dans les 26 semaines précédentes étaient inéligibles. Les patients ont été randomisés (2:1) pour recevoir un des traitements suivants :
· Pembrolizumab 200 mg avec pemetrexed 500 mg/m2et, au choix de l'investigateur, cisplatine 75 mg/m2 ou carboplatine ASC 5 mg/mL/min par voie intraveineuse toutes les 3 semaines pendant 4 cycles, puis pembrolizumab 200 mg et pemetrexed 500 mg/m2 par voie intraveineuse toutes les 3 semaines (n = 410) ;· Placebo avec pemetrexed 500 mg/m2et, au choix de l'investigateur, cisplatine 75 mg/m2 ou carboplatine ASC 5 mg/mL/min par voie intraveineuse toutes les 3 semaines pendant 4 cycles, puis placebo et pemetrexed 500 mg/m2 par voie intraveineuse toutes les 3 semaines (n = 206).
Le traitement par pembrolizumab était poursuivi jusqu'à progression de la maladie définie par RECIST 1.1 telle que déterminée par l'investigateur, toxicité inacceptable ou durée maximale de 24 mois. L'administration de pembrolizumab était autorisée au-delà de la progression de la maladie définie par RECIST et évaluée en aveugle par une revue centralisée indépendante ou au-delà de l'arrêt de pemetrexed si le patient était cliniquement stable et si l'investigateur considérait qu'il y avait un bénéfice clinique. Chez les patients ayant terminé les 24 mois de traitement ou ayant une réponse complète, le traitement par pembrolizumab pouvait être réinitié en cas de progression de la maladie et administré jusqu'à une année supplémentaire. Une évaluation de la réponse tumorale était réalisée à la semaine 6 et à la semaine 12 puis toutes les 9 semaines ensuite. Les patients recevant placebo plus chimiothérapie qui présentaient une progression de leur maladie vérifiée de manière indépendante pouvaient bénéficier de pembrolizumab en monothérapie.
Parmi les 616 patients de KEYNOTE-189, les caractéristiques à l'inclusion étaient : âge médian de 64 ans (49 % âgés de 65 ans ou plus) ; 59 % d'hommes ; 94 % de type caucasien et 3 % de type asiatique ; 43% et 56 % avaient un statut de performance ECOG de 0 ou 1, respectivement ; 31 % étaient PD-L1 négatif (TPS < 1 %) ; et 18 % avaient des métastases cérébrales traitées ou non traitées à l'inclusion. Les critères principaux d'évaluation de l'efficacité étaient l'OS et la PFS (telles qu'évaluées en aveugle par une revue centralisée indépendante utilisant RECIST 1.1). Les critères secondaires d'évaluation de l'efficacité étaient l'ORR et la durée de réponse, telles qu'évaluées en aveugle par une revue centralisée indépendante utilisant RECIST 1.1. Le tableau 14 résume les résultats principaux d'efficacité et les figures 11 et 12 montrent les courbes de Kaplan-Meier pour l'OS et la PFS, basées sur l'analyse finale avec un suivi médian de 18,8 mois.
Tableau 14 : Résultats d'ef icacité dans l'étude KEYNOTE-189
| Critère | Pembrolizumab + Pemetrexed + Chimiothérapie à base de sels de platinen = 410 | Placebo + Pemetrexed +Chimiothérapie à base de sels de platinen = 206 |
| OS* | ||
| Nombre (%) de patients avec événement | 258 (63 %) | 163 (79 %) |
| Hazard ratio† (IC 95%) | 0,56 (0,46 - 0,69) | |
| Valeur de p‡ | < 0,00001 | |
| Médiane en mois (IC 95%) | 22,0(19,5 - 24,5) | 10,6(8,7 - 13,6) |
| PFS | ||
| Nombre (%) de patients avec événement | 337 (82 %) | 197 (96 %) |
| Hazard ratio† (IC 95%) | 0,49 (0,41 - 0,59) | |
| Valeur de p‡ | < 0,00001 | |
| Médiane en mois (IC 95%) | 9,0 (8,1 - 10,4) | 4,9 (4,7 - 5,5) |
| Taux de réponse objective | ||
| ORR§ % (IC 95%) | 48 % (43 - 53) | 20 % (15 - 26) |
| Réponse complète | 1,2 % | 0,5 % |
| Réponse partielle | 47 % | 19 % |
| Valeur de p¶ | < 0,0001 | |
| Durée de réponse | ||
| Médiane en mois (intervalle) | 12,5(1,1+ - 34,9+) | 7,1(2,4 - 27,8+) |
| % avec durée ≥ 12 mois# | 53 % | 27 % |
* Un total de 113 patients (57 %) ayant interrompu le traitement à l'étude dans le bras placebo plus chimiothérapie ont reçu en cross-over pembrolizumab en monothérapie, ou ont reçu un inhibiteur de point de contrôle en traitement ultérieur
† Sur la base du modèle « Cox proportional hazard » stratifié
‡ Sur la base du test de Log rank stratifié
§ Sur la base des patients avec, pour meilleure réponse objective, une réponse complète ou partielle confirmée
¶ Sur la base de la méthode de Miettinen et Nurminen stratifiée par statut PD-L1, chimiothérapie à base de sels de platine et statut tabagique
# Sur la base de l'estimation de Kaplan-Meier
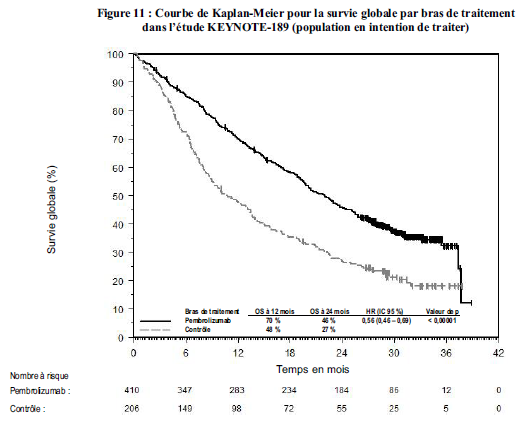
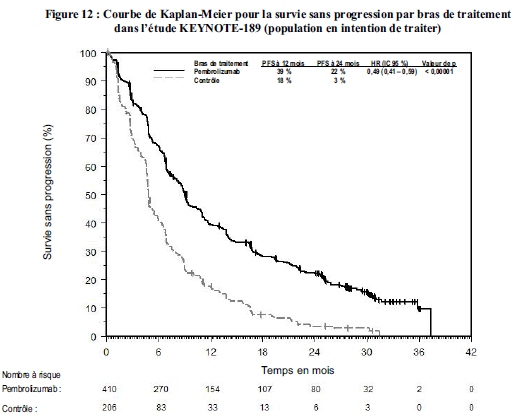
Une analyse a été réalisée dans KEYNOTE-189 chez les patients qui avait un PD-L1 TPS < 1% [pembrolizumab en association : n = 127 (31 %) vs. chimiothérapie : n = 63 (31 %)], TPS 1-49 % pembrolizumab en association: n = 128 (31 %) vs. chimiothérapie : n = 58 (28 %)] ou ≥ 50 % pembrolizumab en association : n = 132 (32 %) vs. chimiothérapie : n = 70 (34 %)] (voir Tableau 15).
Tableau 15 : Résultats d'ef icacité selon l'expression de PD-L1 dans KEYNOTE-189*
| Critère | Pembrolizumab en association | Chimiothérapie | Pembrolizumab en association | Chimiothérapie | Pembrolizumab en association | Chimiothérapie |
| TPS < 1 % | TPS 1 à 49 % | TPS ≥ 50 % | ||||
| OS Hazard ratio† (IC 95 %) | 0,51 (0,36 - 0,71) | 0,66 (0,46 - 0,96) | 0,59 (0,40 - 0,86) | |||
| PFS Hazard ratio†(IC 95 %) | 0,67 (0,49 - 0,93) | 0,53 (0,38 - 0,74) | 0,35 (0,25 - 0,49) | |||
| ORR % | 33 % | 14 % | 50 % | 21 % | 62 % | 26 % |
* Basé sur l'analyse finale
† Hazard ratio (pembrolizumab en association comparé à la chimiothérapie) sur la base du modèle « Cox proportional hazard » stratifié
Lors de l'analyse finale, un total de 57 patients âgés de ≥ 75 ans atteints d'un CBNPC ont été inclus dans l'étude KEYNOTE-189 (35 dans le groupe pembrolizumab en association et 22 dans le groupe contrôle). Un HR = 1,54 [IC 95 % : 0,76 - 3,14] pour l'OS et un HR = 1,12 [IC 95 % : 0,56 - 2,22] pour la PFS pour le pembrolizumab en association vs chimiothérapie a été rapporté dans ce sous-groupe de l'étude. Les données sur l'ef icacité de pembrolizumab en association à la chimiothérapie à base de sels de platine sont limitées dans cette population.
KEYNOTE-407 : Etude contrôlée du traitement en association chez des patients naïfs de traitement atteints d'un CBNPC épidermoïde
L'efficacité de pembrolizumab en association au carboplatine et au paclitaxel ou au nab-paclitaxel a été étudiée dans l'étude KEYNOTE-407, une étude randomisée, en double-aveugle, multicentrique, contrôlée par placebo. Les principaux critères d'éligibilité pour cette étude étaient un CBNPC métastatique épidermoïde, quel que soit le statut d'expression tumorale de PD-L1 et sans traitement systémique antérieur pour la maladie métastatique. Les patients atteints d'une maladie auto-immune ayant nécessité un traitement systémique dans les 2 ans précédents, ceux dont l'état clinique nécessitait un traitement immunosuppresseur ou ceux qui avaient reçu plus de 30 Gy d'irradiation thoracique au cours des 26 semaines précédentes étaient inéligibles. La randomisation était stratifiée selon l'expression tumorale de PD-L1 (TPS < 1 % [négatif] versus TPS ≥ 1 %), le choix de l'investigateur : placlitaxel ou nab-paclitaxel, et la région géographique (Asie de l'Est vs non Asie de l'Est). Les patients étaient randomisés (1:1) dans l'un des bras de traitement suivants, en perfusion intraveineuse :
- Pembrolizumab 200 mg et carboplatine ASC 6 mg/mL/min le jour 1 de chaque cycle de 21 jours pendant 4 cycles, et paclitaxel 200 mg/m2 le jour 1 de chaque cycle de 21 jours pendant 4 cycles ou nab-paclitaxel 100 mg/m2 les jours 1, 8 et 15 de chaque cycle de 21 jours pendant 4 cycles, puis pembrolizumab 200 mg toutes les 3 semaines. Le pembrolizumab était administré avant la chimiothérapie le jour 1.
- Placebo et carboplatine ASC 6 mg/mL/min le jour 1 de chaque cycle de 21 jours pendant 4 cycles, et paclitaxel 200 mg/m2 le jour 1 de chaque cycle de 21 jours pendant 4 cycles ou nab-paclitaxel 100 mg/m2 les jours 1, 8 et 15 de chaque cycle de 21 jours pendant 4 cycles, puis placebo toutes les 3 semaines.
Le traitement par pembrolizumab ou placebo était poursuivi jusqu'à progression de la maladie définie par RECIST 1.1 telle que déterminée en aveugle par une revue centralisée indépendante, toxicité inacceptable, ou durée maximale de 24 mois. L'administration de pembrolizumab était autorisée au-delà de la progression de la maladie définie par RECIST si le patient était cliniquement stable et si l'investigateur considérait qu'il y avait un bénéfice clinique.
Les patients dans le groupe placebo pouvaient bénéficier, au moment de la progression de la maladie, de pembrolizumab en monothérapie.
Une évaluation de la réponse tumorale était réalisée toutes les 6 semaines jusqu'à la semaine 18, toutes les 9 semaines jusqu'à la semaine 45 et toutes les 12 semaines ensuite.
Un total de 559 patients ont été randomisés. Les caractéristiques de la population de l'étude étaient : âge médian de 65 ans (29 à 88 ans) : 55 % âgés de 65 ans ou plus ; 81 % d'hommes, 77 % de type caucasien ; statut de performance ECOG de 0 (29 %) et 1 (71 %) ; et 8 % avec des métastases cérébrales traitées à l'inclusion. Trente-cinq pourcents avaient une expression tumorale de PD-L1 TPS < 1 % (négatif) ; 19 % étaient originaires d'Asie de l'Est ; et 60 % ont reçu du paclitaxel.
Les critères principaux d'évaluation de l'efficacité étaient l'OS et la PFS (telles qu'évaluées en aveugle par une revue centralisée indépendante utilisant RECIST 1.1). Les critères secondaires d'évaluation de l'efficacité étaient l'ORR et la durée de réponse, telles qu'évaluées en aveugle par une revue centralisée indépendante utilisant RECIST 1.1. Le tableau 16 résume les critères clés d'efficacité et les figures 13 et 14 montrent les courbes de Kaplan-Meier pour l'OS et la PFS, basées sur l'analyse finale avec un suivi médian de 14,3 mois.
Tableau 16 : Résultats d'ef icacité dans l'étude KEYNOTE-407
| Critère d'évaluation | Pembrolizumab Carboplatine Paclitaxel/Nab-paclitaxel n = 278 | Placebo Carboplatine Paclitaxel/Nab-paclitaxel n = 281 |
| OS* | ||
| Nombre (%) de patients avec événement | 168 (60 %) | 197 (70 %) |
| Médiane en mois (IC 95 %) | 17,1 (14,4 - 19,9) | 11,6 (10,1 - 13,7) |
| Hazard ratio† (IC 95 %) | 0,71 (0,58 - 0,88) | |
| Valeur de p‡ | 0,0006 | |
| PFS | ||
| Nombre (%) de patients avec événement | 217 (78 %) | 252 (90 %) |
| Médiane en mois (IC 95 %) | 8,0 (6,3 - 8,4) | 5,1 (4,3 - 6,0) |
| Hazard ratio† (IC 95 %) | 0,57 (0,47 - 0,69) | |
| Valeur de p‡ | < 0,0001 | |
| Taux de réponse objective | ||
| ORR % (IC 95 %) | 63 % (57 - 68) | 38 % (33 - 44) |
| Réponse complète | 2,2 % | 3,2 % |
| Réponse partielle | 60 % | 35 % |
| Valeur de p§ | < 0,0001 | |
| Durée de réponse | ||
| Médiane en mois (intervalle) | 8,8 (1,3+ - 28,4+) | 4,9 (1,3+ - 28,3+) |
| % avec durée ≥ 12 mois¶ | 38 % | 25 % |
† Sur la base du modèle « Cox proportional hazard » stratifié
‡ Sur la base du test de log rank stratifié
§ Sur la base de la méthode de Miettinen et Nurminen
¶ Sur la base de l'estimation de Kaplan-Meier
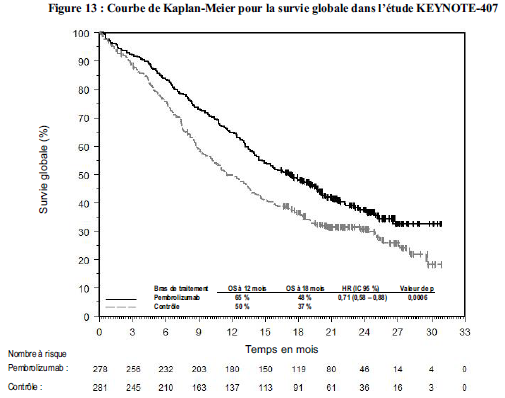
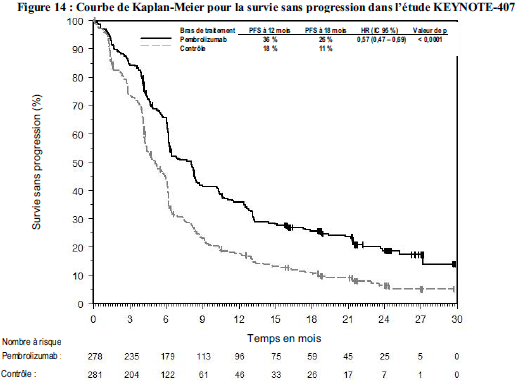
Une analyse a été réalisée dans KEYNOTE-407 chez les patients qui avaient un PD-L1 TPS < 1 % [bras pembrolizumab plus chimiothérapie : n = 95 (34 %) vs. bras placebo plus chimiothérapie : n = 99 (35 %)], TPS 1 % à 49 % [bras pembrolizumab plus chimiothérapie : n = 103 (37 %) vs. bras placebo plus chimiothérapie : n = 104 (37 %)] ou TPS ≥ 50 % [bras pembrolizumab plus chimiothérapie : n = 73 (26 %) vs. bras placebo plus chimiothérapie : n = 73 (26 %)] (voir Tableau 17).
Tableau 17 : Résultats d'ef icacité selon l'expression de PD-L1 dans KEYNOTE-407*
| Critèred'évaluation | Pembrolizumab en association | Chimiothérapie | Pembrolizumab en association | Chimiothérapie | Pembrolizumab en association | Chimiothérapie |
| TPS < 1 % | TPS 1 à 49 % | TPS ≥ 50 % | ||||
| OS Hazard ratio†(IC 95 %) | 0,79 (0,56 - 1,11) | 0,59 (0,42 - 0,84) | 0,79 (0,52 - 1,21) | |||
| PFS Hazard ratio†(IC 95 %) | 0,67 (0,49 - 0,91) | 0,52 (0,38 - 0,71) | 0,43 (0,29 - 0,63) | |||
| ORR % | 67 % | 41 % | 55 % | 42 % | 64 % | 30 % |
† Hazard ratio (pembrolizumab en association compare à la chimiothérapie) sur la base du modèle « Cox proportional hazard » stratifié
Lors de l'analyse finale, un total de 65 patients âgés de ≥ 75 ans atteints d'un CBNPC ont été inclus dans l'étude KEYNOTE-407 (34 dans le groupe pembrolizumab en association et 31 dans le groupe contrôle). Un HR = 0,81 [IC 95 % : 0,43 - 1,55] pour l'OS, un HR = 0,61 [IC 95 % : 0,34 - 1,09] pour la PFS et des ORR de 62 % et 45 % pour le pembrolizumab en association vs chimiothérapie ont été rapportés dans ce sous-groupe de l'étude. Les données sur l'ef icacité de pembrolizumab en association à la chimiothérapie à base de sels de platine sont limitées dans cette population de patients.
KEYNOTE-010 : Etude contrôlée chez des patients atteints d'un CBNPC précédemment traités par chimiothérapie
L'efficacité et la tolérance de pembrolizumab ont été étudiées dans KEYNOTE-010, une étude multicentrique, en ouvert, contrôlée, dans le traitement des patients atteints d'un CBNPC avancé prétraités par une chimiothérapie à base de sels de platine. Les patients devaient exprimer PD-L1 avec un TPS ≥1 % avec le kit PD-L1 IHC 22C3 pharmDxTM. Avant de recevoir pembrolizumab, les patients présentant une mutation activatrice d'EGFR ou une translocation d'ALK devaient aussi avoir une progression de leur maladie sous un traitement autorisé pour ces mutations. Les patients ont été randomisés (1:1:1) pour recevoir pembrolizumab à la dose de 2 mg/kg (n = 344) ou 10 mg/kg de poids corporel (n = 346) toutes les 3 semaines ou docétaxel à la dose de 75 mg/m2 toutes les 3 semaines (n = 343) jusqu'à progression de la maladie ou toxicité inacceptable. L'étude excluait les patients atteints d'une maladie auto-immune ou ceux dont l'état clinique nécessitait un immunosuppresseur ou ceux qui avaient reçu plus de 30 Gy d'irradiation thoracique dans les 26 semaines précédentes. Une évaluation de la réponse tumorale était réalisée toutes les 9 semaines.
Les caractéristiques à l'inclusion pour cette population comprenaient : âge médian de 63 ans (42 % âgés de 65 ans ou plus), 61 % d'hommes, 72 % de type caucasien et 21 % de type asiatique. Le statut de performance ECOG était de 0 et 1 chez 34 % et 66 % des patients, respectivement. Les caractéristiques de la maladie étaient : histologie épidermoïde (21 %) et non-épidermoïde (70 %) ; stade IIIA (2 %) ; stade IIIB (7 %) ; stade IV (91 %) ; métastases cérébrales stables (15 %) ; incidence des mutations : EGFR (8 %) ou ALK (1 %). Le traitement antérieur comprenait un doublet à base de sels de platine (100 %) ; les patients avaient reçu une (69 %), ou au moins deux (29 %) lignes de traitement.
Les critères principaux d'évaluation de l'ef icacité étaient l'OS et la PFS telles qu'évaluées en aveugle par une revue centralisée indépendante utilisant RECIST 1.1. Les critères secondaires d'évaluation d'ef icacité étaient l'ORR et la durée de réponse. Le tableau 18 résume les critères clés d'ef icacité pour la population entière (TPS ≥ 1 %) et pour la population de patients avec TPS ≥ 50 % et la figure 15 montre la courbe de Kaplan-Meier pour l'OS (TPS ≥ 1 %), basée sur une analyse finale avec un suivi médian de 42,6 mois.
Tableau 18 : Réponse à pembrolizumab à 2 ou 10 mg/kg de poids corporel toutes les 3 semaines chez les patients précédemment traités atteints d'un CBNPC dans l'étude KEYNOTE-010
| Critère d'évaluation | pembrolizumab 2 mg/kg de poids corporel toutes les 3 semaines | pembrolizumab 10 mg/kg de poids corporel toutes les3 semaines | docétaxel75 mg/m2 toutesles 3 semaines |
| TPS ≥ 1% | |||
| Nombre de patients | 344 | 346 | 343 |
| OS | |||
| Nombre (%) de patients avec évènement | 284 (83 %) | 264 (76 %) | 295 (86 %) |
| Hazard ratio* (IC 95 %) | 0,77 (0,66 - 0,91) | 0,61 (0,52 - 0,73) | --- |
| Valeur de p† | 0,00128 | < 0,001 | --- |
| Médiane en mois (IC 95 %) | 10,4 (9,5 - 11,9) | 13,2 (11,2 - 16,7) | 8,4 (7,6 - 9,5) |
| PFS‡ | |||
| Nombre (%) de patients avec évènement | 305 (89 %) | 292 (84%) | 314 (92 %) |
| Hazard ratio* (IC 95 %) | 0,88 (0,75 - 1,04) | 0,75 (0,63 - 0,89) | --- |
| Valeur de p† | 0,065 | < 0,001 | --- |
| Médiane en mois (IC 95 %) | 3,9 (3,1 - 4,1) | 4,0 (2,7 - 4,5) | 4,1 (3,8 - 4,5) |
| Taux de réponse objective‡ | |||
| ORR % (IC 95 %) | 20 % (16 - 25) | 21 % (17 - 26) | 9 % (6 - 13) |
| Réponse complète | 2 % | 3 % | 0 % |
| Réponse partielle | 18 % | 18 % | 9 % |
| Critère d'évaluation | pembrolizumab 2 mg/kg de poids corporel toutes les 3 semaines | pembrolizumab 10 mg/kg de poids corporel toutes les3 semaines | docétaxel75 mg/m2 toutesles 3 semaines |
| Durée de réponse ‡,§ | |||
| Médiane en mois (intervalle) | Non atteinte(2,8 - 46,2+) | 37,8(2,0+ - 49,3+) | 7,1(1,4+ - 16,8) |
| % en cours¶ | 42 % | 43 % | 6 % |
| TPS ≥ 50 % | |||
| Nombre de patients | 139 | 151 | 152 |
| OS | |||
| Nombre (%) de patients avec évènement | 97 (70 %) | 102 (68 %) | 127 (84 %) |
| Hazard ratio* (IC 95 %) | 0,56 (0,43 - 0,74) | 0,50 (0,38 - 0,65) | --- |
| Valeur de p † | < 0,001 | < 0,001 | --- |
| Médiane en mois (IC 95 %) | 15,8 (10,8 - 22,5) | 18,7 (12,1 -25,3) | 8,2 (6,4 - 9,8) |
| PFS‡ | |||
| Nombre (%) de patients avec évènement | 107 (77 %) | 115 (76 %) | 138 (91 %) |
| Hazard ratio* (IC 95 %) | 0,59 (0,45 - 0,77) | 0,53 (0,41 - 0,70) | --- |
| Valeur de p† | < 0,001 | < 0,001 | --- |
| Médiane en mois (IC 95 %) | 5,3 (4,1 - 7,9) | 5,2 (4,1 - 8,1) | 4,2 (3,8 - 4,7) |
| Taux de réponse objective‡ | |||
| ORR % (IC 95 %) | 32 % (24 - 40) | 32 % (25 - 41) | 9 % (5 - 14) |
| Réponse complète | 4 % | 4 % | 0 % |
| Réponse partielle | 27 % | 28 % | 9 % |
| Durée de réponse‡,§ | |||
| Médiane en mois (intervalle) | Non atteinte(2,8 - 44,0+) | 37,5(2,0+ - 49,3+) | 8,1(2,6+ - 16,8) |
| % en cours¶ | 55 % | 47 % | 8 % |
† Sur la base du test de log rank stratifié
‡ Evaluation en aveugle par une revue centralisée indépendante utilisant RECIST 1.1
§ Sur la base des patients avec, pour meilleure réponse objective, une réponse complète ou partielle confirmée
¶ Réponse en cours inclut tous les répondeurs qui, au moment de l'analyse, étaient vivants, sans progression, n'avaient pas débuté de nouveaux traitements anticancéreux et n'avaient pas été définis comme perdus de vue pour le suivi
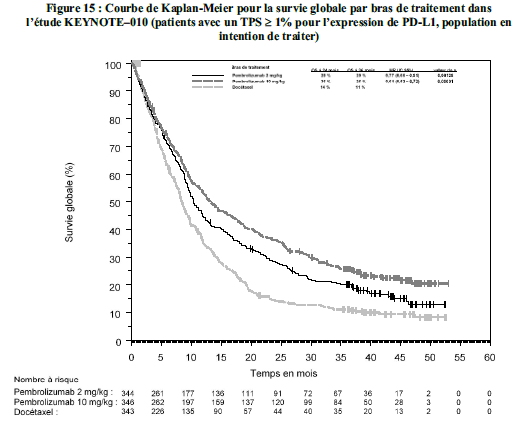
Les résultats d'efficacité ont été similaires pour les bras pembrolizumab 2 mg/kg de poids corporel et 10 mg/kg de poids corporel. Les résultats d'ef icacité pour la survie globale ont été cohérents quelle que soit l'ancienneté de la biopsie tumorale (fraiche vs archivée) sur la base d'une comparaison inter-groupe.
Dans des analyses en sous-groupes, le bénéfice de survie observé avec le pembrolizumab par rapport au docétaxel a été moindre chez les patients n'ayant jamais fumé, ou chez les patients présentant des tumeurs avec mutations activatrices d'EGFR qui avaient reçu au moins une chimiothérapie à base de sels de platine et un inhibiteur de tyrosine-kinase ; cependant, aucune conclusion définitive ne peut être tirée de ces données en raison du petit nombre de patients.
L'efficacité et la tolérance du pembrolizumab chez les patients dont la tumeur n'exprime pas PD-L1 n'ont pas pu être établies.
Lymphome de Hodgkin classique KEYNOTE-204 : Etude contrôlée chez des patients atteints d'un lymphome de Hodgkin classique (LHc) en rechute ou réfractaire
L'efficacité du pembrolizumab a été étudiée dans KEYNOTE-204, une étude randomisée, en ouvert, contrôlée par traitement actif, menée chez 304 patients atteints d'un LHc en rechute ou réfractaire. Les patients avec une pneumopathie non infectieuse active, une GCSH allogénique dans les 5 dernières années (ou > 5 ans mais avec des symptômes de GVH), une maladie auto-immune active, un état clinique ayant nécessité un traitement immunosuppresseur ou une infection active nécessitant un traitement systémique étaient inéligibles à l'étude. La randomisation était stratifiée selon la présence d'une GCS autologue antérieure (oui vs non) et le statut de la maladie après un traitement de première intention (réfractaire primaire vs rechute moins de 12 mois après la fin du traitement vs rechute 12 mois ou plus après la fin du traitement). Les patients étaient randomisés (1:1) dans l'un des bras de traitement suivants :
- Pembrolizumab 200 mg par voie intraveineuse toutes les 3 semaines
- Brentuximab vedotin (BV) 1,8 mg/kg de poids corporel par voie intraveineuse toutes les 3 semaines.
Les patients ont reçu 200 mg de pembrolizumab par voie intraveineuse toutes les 3 semaines jusqu'à toxicité inacceptable ou progression documentée de la maladie ou un maximum de 35 cycles. Des données limitées sont actuellement disponibles concernant la durée de réponse après l'arrêt du pembrolizumab à 35 cycles. La réponse était évaluée toutes les 12 semaines, avec la première évaluation post-inclusion planifiée à la Semaine 12.
Parmi les 304 patients de l'étude KEYNOTE-204, il existe une sous-population composée de 112 patients en échec d'une greffe avant l'inclusion et 137 patients en échec d'au moins 2 lignes de traitement antérieures et inéligibles à une GCS autologue au moment de l'inclusion. Les caractéristiques à l'inclusion de ces 249 patients étaient : âge médian de 34 ans (11 % âgés de 65 ans ou plus), 56 % d'hommes, 80 % de type caucasien et 7 % de type asiatique et le statut de performance ECOG était de 0 et 1 pour 58 % et 41 % des patients, respectivement. Approximativement 30 % des patients étaient réfractaires à une chimiothérapie de première intention et ~ 45 % avaient eu une GCS autologue antérieure. Le sous-type histologique de LHc le plus représenté (~ 81%) était scléro-nodulaire et une masse tumorale volumineuse, des symptômes B et un envahissement de la moelle osseuse étaient présents chez approximativement 21 %, 28 % et 4 % des patients, respectivement.
Le critère principal d'évaluation de l'efficacité était la PFS et le critère secondaire d'évaluation de l'efficacité était l'ORR, tous les deux évalués en aveugle par une revue centralisée indépendante selon les critères de l'International Working Group (IWG) révisés en 2007. Le critère principal additionnel d'efficacité, l'OS, n'a pas été évalué formellement au moment de l'analyse. Dans la population en intention de traiter, le suivi médian était de 24,9 mois (de 1,8 à 42,0 mois) pour les 151 patients traités par pembrolizumab. L'analyse initiale a abouti à un HR pour la PFS de 0,65 (IC 95 % ; 0,48 - 0,88) avec une valeur de p unilatérale de 0,0027. L'ORR était de 66 % pour le pembrolizumab comparé à 54 % pour le traitement standard avec une valeur de p de 0,0225. Le tableau 19 résume les résultats d'efficacité dans la sous-population. Les résultats d'efficacité dans cette sous-population étaient cohérents avec la population en intention de traiter. La figure 16 représente la courbe de Kaplan-Meier pour la PFS pour cette sous- population.
Tableau 19 : Résultats d'efficacité chez les patients atteints d'un LHc en échec d'une greffe avant l'inclusion ou en échec d'au moins 2 lignes de traitement antérieures et inéligibles à une GCS autologue dans l'étude KEYNOTE-204
| Critère d'évaluation | Pembrolizumab200 mg toutes les 3 semainesn = 124 | Brentuximab vedotin1,8 mg/kg de poids corporel toutes les 3 semainesn = 125 |
| PFS | ||
| Nombre (%) de patients avec évènement | 68 (55 %) | 75 (60 %) |
| Hazard ratio* (IC 95 %) | 0,66 (0,47 - 0,92) | |
| Médiane en mois (IC 95 %) | 12,6 (8,7 - 19,4) | 8,2 (5,6 - 8,8) |
| Taux de réponse objective | ||
| ORR‡ % (IC 95 %) | 65 % (56,3 - 73,6) | 54 % (45,3 - 63,3) |
| Réponse complète | 27 % | 22 % |
| Réponse partielle | 39 % | 33 % |
| Maladie stable | 12 % | 23 % |
| Durée de réponse | ||
| Médiane en mois (intervalle) | 20,5 (0,0+ - 33,2+) | 11,2 (0,0+ - 33,9+) |
| Nombre (%¶) de patients avec durée ≥ 6 mois | 53 (80,8 %) | 28 (61,2 %) |
| Nombre (%¶) de patients avec durée ≥ 12 mois | 37 (61,7 %) | 17 (49,0 %) |
‡ Sur la base des patients avec, pour meilleure réponse objective, une réponse complète ou partielle
¶ Sur la base de l'estimation de Kaplan-Meier

KEYNOTE-087 et KEYNOTE-013 : Etudes en ouvert chez des patients atteints d'un LHc en rechute ou réfractaires
L'efficacité du pembrolizumab a été étudiée dans KEYNOTE-087 et KEYNOTE-013, deux études multicentriques en ouvert dans le traitement de 241 patients atteints d'un LHc. Ces études ont inclus des patients en échec d'une GCS autologue et d'un traitement par BV ; des patients inéligibles à une GCS autologue du fait de l'absence de rémission complète ou partielle avec une chimiothérapie de rattrapage, et en échec de BV ; ou des patients en échec d'une GCS autologue et n'ayant pas reçu de BV. Cinq sujets de l'étude étaient inéligibles à une GCS autologue pour d'autres raisons que l'échec de la chimiothérapie de rattrapage. Les deux études incluaient les patients indépendamment de l'expression PD-L1. Les patients avec une pneumopathie non infectieuse active, avec une greffe allogénique dans les 5 dernières années (ou > 5 ans mais avec GVH), avec une maladie auto-immune active ou avec un état clinique ayant nécessité un traitement immunosuppresseur, étaient inéligibles à ces deux études. Les patients ont reçu 200 mg de pembrolizumab toutes les 3 semaines (n = 210 : KEYNOTE-087) ou 10 mg/kg de poids corporel toutes les 2 semaines (n = 31 : KEYNOTE-013) jusqu'à toxicité inacceptable ou progression documentée de la maladie.
Parmi les patients de l'étude KEYNOTE-087, les caractéristiques à l'inclusion étaient : âge médian de 35 ans (9 % âgés de 65 ans ou plus), 54 % d'hommes, 88 % de type caucasien et le statut de performance ECOG était de 0 et 1 chez 49 % et 51 % des patients, respectivement. Le nombre médian de lignes de traitement antérieures administrées pour traiter le LHc était de 4 (de 1 à 12). Quatre-vingt-un pourcent étaient réfractaires à au moins un traitement préalable dont 34 % réfractaires à la première ligne de traitement. Soixante-et-un pourcent des patients avaient reçu une GCS autologue, 38 % étaient inéligibles à la greffe, 17 % n'avaient pas eu d'administration préalable de brentuximab vedotin et 37 % des patients avaient reçu une radiothérapie préalable. Les sous-types de la maladie étaient : 81 % scléro-nodulaire, 11 % à cellularité mixte, 4 % riche en lymphocytes et 2 % à déplétion lymphocytaire.
Parmi les patients de l'étude KEYNOTE-013, les caractéristiques à l'inclusion étaient : âge médian de 32 ans (7 % âgés de 65 ans ou plus), 58 % d'hommes, 94 % de type caucasien et le statut de performance ECOG était de 0 et 1 chez 45 % et 55 % des patients, respectivement. Le nombre médian de lignes de traitement antérieures administrées pour traiter le LHc était de 5 (de 2 à 15). Quatre-vingt-quatre pourcent étaient réfractaires à au moins un traitement préalable dont 35 % réfractaires à la première ligne de traitement. Soixante-quatorze pourcent des patients avaient reçu une GCS autologue, 26 % étaient inéligibles à la greffe et 45 % des patients avaient reçu une radiothérapie préalable. Les sous-types de la maladie étaient : 97 % scléro-nodulaire et 3 % à cellularité mixte.
Les critères principaux d'évaluation de l'ef icacité (taux de réponse objective - ORR et taux de réponse complète - CRR) ont été évalués en aveugle par une revue centralisée indépendante selon les critères de l'IWG de 2007. Les critères secondaires d'ef icacité étaient la durée de réponse, la PFS et l'OS. La réponse était évaluée dans les études KEYNOTE-087 et KEYNOTE-013 toutes les 12 semaines et 8 semaines, respectivement, avec la première évaluation post-inclusion planifiée à la Semaine 12. Les principaux résultats d'efficacité sont résumés dans le tableau 20.
Tableau 20 : Résultats d'ef icacité dans les études KEYNOTE-087 et KEYNOTE-013
| KEYNOTE-087* | KEYNOTE-013† | |
| Critère d'évaluation | Pembrolizumab 200 mg toutes les 3 semaines n = 210 | Pembrolizumab10 mg/kg de poids corporel toutes les 2 semaines n = 31 |
| Taux de réponse objective‡ | ||
| ORR % (IC 95 %) | 71 % (64,8 - 77,4) | 58 % (39,1 - 75,5) |
| Rémission complète | 28 % | 19 % |
| Rémission partielle | 44 % | 39 % |
| Durée de réponse‡ | ||
| Médiane en mois (intervalle) | 16,6 (0,0+ - 62,1+)§ | Non atteinte (0,0+ - 45,6+)¶ |
| % avec durée ≥ 12 mois | 59 %# | 70 %Þ |
| % avec durée ≥ 24 mois | 45 %ß | --- |
| % avec durée ≥ 60 mois | 25 %à | --- |
| Délai de réponse | ||
| Médiane en mois (intervalle) | 2,8 (2,1 - 16,5)§ | 2,8 (2,4 - 8,6)¶ |
| OS | ||
| Nombre (%) de patients avec évènement | 59 (28 %) | 6 (19 %) |
| OS à 12 mois | 96 % | 87 % |
| OS à 24 mois | 91 % | 87 % |
| OS à 60 mois | 71 % | --- |
† Suivi médian de 52,8 mois
‡ Evalués en aveugle par une revue centralisée indépendante en utilisant les critères de l'IWG de 2007 par scanner PET/CT
§ Sur la base des patients (n = 150) avec une réponse confirmée par une revue indépendante
¶ Sur la base des patients (n = 18) avec une réponse confirmée par une revue indépendante
# Sur la base de l'estimation de Kaplan-Meier ; inclut 62 patients avec des réponses de 12 mois ou plus Þ Sur la base de l'estimation de Kaplan-Meier ; inclut 7 patients avec des réponses de 12 mois ou plus ß Sur la base de l'estimation de Kaplan-Meier ; inclut 37 patients avec des réponses de 24 mois ou plus à Sur la base de l'estimation de Kaplan-Meier ; inclut 4 patients avec des réponses de 60 mois ou plus
Efficacité chez les patients âgés
Au total, 46 patients âgés de ≥ 65 ans avec un LHc ont été traités par pembrolizumab dans les études KEYNOTE-087, KEYNOTE-013 et KEYNOTE-204. Les données chez ces patients sont trop limitées pour tirer une conclusion en termes d'efficacité dans cette population.
Carcinome urothélial
KEYNOTE-045 : Etude contrôlée chez des patients atteints d'un carcinome urothélial ayant reçu une
chimiothérapie antérieure à base de sels de platine
L'ef icacité et la tolérance de pembrolizumab ont été évaluées dans l'étude KEYNOTE-045, une étude multicentrique, en ouvert, randomisée (1:1), contrôlée, dans le traitement du carcinome urothélial localement avancé ou métastatique chez des patients qui présentaient une progression de la maladie pendant ou après une chimiothérapie à base de sels de platine. Les patients devaient avoir reçu une première ligne de traitement à base de sels de platine pour une maladie localement avancée/métastatique ou comme traitement néoadjuvant/adjuvant, avec récidive/progression dans un délai ≤ 12 mois après la fin du traitement. Les patients ont été randomisés (1:1) pour recevoir soit pembrolizumab 200 mg toutes les 3 semaines (n = 270), soit, au choix de l'investigateur, l'une des chimiothérapies suivantes, toutes administrées par voie intraveineuse toutes les 3 semaines (n = 272) : paclitaxel 175 mg/m2 (n = 84), docétaxel 75 mg/m2 (n = 84) ou vinflunine 320 mg/m2 (n = 87). Les patients étaient traités par pembrolizumab jusqu'à toxicité inacceptable ou progression de la maladie. Le traitement pouvait être poursuivi au-delà de la progression de la maladie si le patient était cliniquement stable et si l'investigateur considérait qu'il y avait un bénéfice clinique. Les patients sans progression de leur maladie pouvaient être traités jusqu'à 24 mois. L'étude excluait les patients atteints d'une maladie auto-immune, les patients dont l'état clinique nécessitait un traitement immunosuppresseur et les patients ayant reçu plus de 2 lignes antérieures de chimiothérapie systémique pour leur carcinome urothélial métastatique. Les patients avec un statut de performance ECOG de 2 devaient avoir une hémoglobine ≥ 10 g/dL, ne pouvaient pas avoir de métastases hépatiques et devaient avoir reçu la dernière dose de leur dernière chimiothérapie précédente au moins 3 mois avant leur inclusion. Une évaluation de la réponse tumorale était réalisée à 9 semaines après la première dose, puis toutes les 6 semaines la première année et ensuite toutes les 12 semaines.
Parmi les 542 patients randomisés dans l'étude KEYNOTE-045, les caractéristiques à l'inclusion étaient : âge médian de 66 ans (de 26 à 88 ans), 58 % âgés de 65 ans ou plus, 74 % d'hommes, 72 % de type caucasien et 23 % de type asiatique, 56 % avaient un statut de performance ECOG de 1 et 1 % avait un statut de performance ECOG de 2, et 96 % présentaient un stade M1 de la maladie et 4 % un stade M0. Quatre- vingt-sept pourcent des patients avaient des métastases viscérales, dont 34 % des métastases hépatiques.
Quatre-vingt-six pourcent des patients avaient une tumeur primitive dans les voies urinaires basses et 14 % dans les voies urinaires hautes. Quinze pourcent des patients présentaient une progression de la maladie suite à une chimiothérapie antérieure néoadjuvante ou adjuvante à base de sels de platine. Vingt-et-un pourcent des patients avaient reçu 2 traitements antérieurs systémiques au stade métastatique. Soixante-seize pourcent des patients avaient reçu un traitement antérieur à base de cisplatine, 23 % à base de carboplatine et 1 % avait été traité avec d'autres traitements à base de sels de platine.
Les critères principaux d'évaluation de l'ef icacité étaient l'OS et la PFS, évaluées en aveugle par une revue centralisée indépendante utilisant RECIST v1.1. Les critères secondaires d'évaluation étaient l'ORR (tel qu'évalué en aveugle par une revue centralisée indépendante utilisant RECIST v1.1) et la durée de réponse. Le tableau 21 résume les critères clés d'ef icacité pour la population en intention de traiter lors de l'analyse finale. La courbe de Kaplan-Meier pour l'OS, basée sur l'analyse finale, est représentée en Figure 17.
L'étude a montré des améliorations statistiquement significatives de l'OS et de l'ORR chez les patients randomisés du groupe pembrolizumab en comparaison à la chimiothérapie. Il n'y avait pas de dif érence statistiquement significative entre le pembrolizumab et la chimiothérapie en ce qui concerne la PFS.
Tableau 21 : Réponse à pembrolizumab 200 mg toutes les 3 semaines chez les patients atteints d'un carcinome urothélial précédemment traités par chimiothérapie dans l'étude KEYNOTE-045
| Critère d'évaluation | Pembrolizumab 200 mg toutes les 3 semainesn = 270 | Chimiothérapien = 272 |
| OS | ||
| Nombre (%) de patients avec événement | 200 (74 %) | 219 (81 %) |
| Hazard ratio* (IC 95 %) | 0,70 (0,57 - 0,85) | |
| Valeur de p† | < 0,001 | |
| Médiane en mois (IC 95 %) | 10,1 (8,0 - 12,3) | 7,3 (6,1 - 8,1) |
| PFS‡ | ||
| Nombre (%) de patients avec événement | 233 (86 %) | 237 (87 %) |
| Hazard ratio* (IC 95 %) | 0,96 (0,79 - 1,16) | |
| Valeur de p† | 0,313 | |
| Médiane en mois (IC 95 %) | 2,1 (2,0 - 2,2) | 3,3 (2,4 - 3,6) |
| Taux de réponse objective‡ | ||
| ORR % (IC 95 %) | 21 % (16 - 27) | 11 % (8 - 15) |
| Valeur de p§ | < 0,001 | |
| Réponse complète | 9 % | 3 % |
| Réponse partielle | 12 % | 8 % |
| Maladie stable | 17 % | 34 % |
| Durée de réponse‡,¶ | ||
| Médiane en mois (intervalle) | Non atteinte (1,6+ - 30,0+) | 4,4(1,4+ - 29,9+) |
| Nombre (%#) de patients avec durée ≥ 6 mois | 46 (84 %) | 8 (47 %) |
| Nombre (%#) de patients avec durée ≥ 12 mois | 35 (68 %) | 5 (35 %) |
† Sur la base du test de log rank stratifié
‡ Evaluation en aveugle par une revue centralisée indépendante utilisant RECIST 1.1
§ Sur la base de la méthode de Miettinen et Nurminen
¶ Sur la base des patients avec, pour meilleure réponse objective, une réponse complète ou partielle confirmée
# Sur la base des estimations de Kaplan-Meier
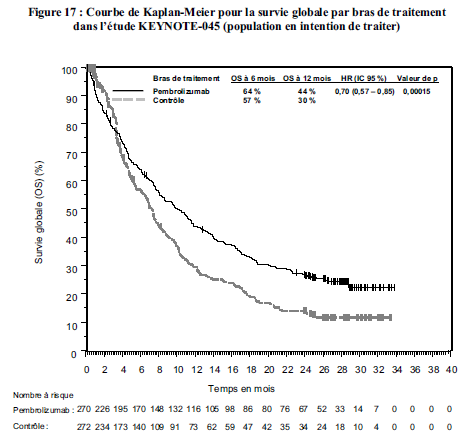
Une analyse a été réalisée dans l'étude KEYNOTE-045 chez des patients qui avaient un CPS de l'expression de PD-L1 < 10 [pembrolizumab : n = 186 (69 %) vs chimiothérapie : n = 176 (65 %)] ou ≥ 10 [pembrolizumab : n = 74 (27 %) vs chimiothérapie : n = 90 (33 %)] dans les deux bras de traitement : pembrolizumab et chimiothérapie (voir tableau 22).
Tableau 22 : OS selon l'expression de PD-L1
| Expression dePD-L1 | Pembrolizumab | Chimiothérapie | |
| OS selon l'expression de PD-L1 Nombre (%) de patients avec événement* | Hazard Ratio† (IC 95 %) | ||
| CPS < 10 | 140 (75 %) | 144 (82 %) | 0,75 (0,59 - 0,95) |
| CPS ≥ 10 | 53 (72 %) | 72 (80 %) | 0,55 (0,37 - 0,81) |
† Hazard ratio (pembrolizumab comparé à la chimiothérapie) sur la base du modèle « Cox proportional hazard » stratifié
Les résultats rapportés par les patients (RRP) ont été évalués à l'aide du questionnaire EORTC QLQ-C30. Il a été observé une prolongation du délai jusqu'à la détérioration de l'état de santé global/qualité de vie dans le questionnaire EORTC QLQ-C30 chez les patients traités par pembrolizumab en comparaison à la chimiothérapie au choix de l'investigateur (HR = 0,70 ; IC 95 % : 0,55 - 0,90). Sur les 15 semaines de suivi, les patients traités par pembrolizumab présentaient un état de santé global/qualité de vie stables alors que ceux traités par une chimiothérapie au choix de l'investigateur avaient un état de santé global/qualité de vie détériorés. Ces résultats sont à interpréter dans le contexte du design de l'étude en ouvert, et doivent par conséquent être pris avec précaution.
KEYNOTE-052 : Etude en ouvert chez des patients atteints d'un carcinome urothélial, inéligibles à une chimiothérapie à base de cisplatine
L'efficacité et la tolérance de pembrolizumab ont été évaluées dans l'étude KEYNOTE-052, une étude multicentrique, en ouvert, dans le traitement du carcinome urothélial localement avancé ou métastatique chez des patients non éligibles à une chimiothérapie à base de cisplatine. Les patients ont reçu pembrolizumab à la dose de 200 mg toutes les 3 semaines jusqu'à toxicité inacceptable ou progression de la maladie. Le traitement pouvait être poursuivi au-delà de la progression de la maladie si le patient était cliniquement stable et si l'investigateur considérait qu'il y avait un bénéfice clinique. Les patients sans progression de leur maladie pouvaient être traités jusqu'à 24 mois. L'étude excluait les patients atteints d'une maladie auto- immune ou les patients dont l'état clinique nécessitait un traitement immunosuppresseur. Une évaluation de la réponse tumorale était réalisée 9 semaines après la première dose, puis toutes les 6 semaines la première année et ensuite toutes les 12 semaines.
Parmi les 370 patients atteints d'un carcinome urothélial qui n'étaient pas éligibles à une chimiothérapie à base de cisplatine, les caractéristiques à l'inclusion étaient : âge médian de 74 ans (82 % âgés de 65 ans ou plus), 77 % d'hommes, 89 % de type caucasien et 7 % de type asiatique. Quatre-vingt-huit pourcent présentaient un stade M1 de la maladie et 12 % un stade M0. Quatre-vingt-cinq pourcent des patients avaient des métastases viscérales dont 21 % des métastases hépatiques. Les raisons de l'inéligibilité au cisplatine incluaient : clairance de la créatinine à l'inclusion < 60 mL/min (50 %), statut de performance ECOG de 2 (32 %), statut de performance ECOG de 2 et clairance de la créatinine à l'inclusion < 60 mL/min (9 %), et autres raisons (insuffisance cardiaque de classe III, neuropathie périphérique de grade 2 ou plus et perte auditive de grade 2 ou plus : 9 %). Quatre-vingt-dix pourcent des patients étaient naïfs de traitement et 10 % avaient reçu une chimiothérapie adjuvante ou néoadjuvante antérieure à base de sels de platine. Quatre-vingt- un pourcent des patients avaient une tumeur primitive dans les voies urinaires basses et 19 % dans les voies urinaires hautes.
Le critère principal d'évaluation de l'efficacité était l'ORR tel qu'évalué en aveugle par une revue centralisée indépendante utilisant RECIST v1.1. Les critères secondaires d'évaluation de l'efficacité étaient la durée de réponse, la PFS et l'OS. Le tableau 23 résume les critères clés d'efficacité pour la population de l'étude lors de l'analyse finale sur la base d'un suivi médian de 11,4 mois (0,1 - 41,2 mois) pour tous les patients.
Tableau 23 : Réponse à pembrolizumab 200 mg toutes les 3 semaines chez les patients atteints d'un carcinome urothélial, inéligibles à une chimiothérapie à base de cisplatine dans l'étude KEYNOTE- 052
| Critère d'évaluation | n = 370 |
| Taux de réponse objective* | |
| ORR %, (IC 95 %) | 29 % (24 - 34) |
| Taux de contrôle de la maladie† | 47 % |
| Réponse complète | 9 % |
| Réponse partielle | 20 % |
| Maladie stable | 18 % |
| Durée de réponse | |
| Médiane en mois (intervalle) | 30,1(1,4+ - 35,9+) |
| % avec durée ≥ 6 mois | 81 %‡ |
| Délai de réponse | |
| Médiane en mois (intervalle) | 2,1 (1,3 - 9,0) |
| Critère d'évaluation | n = 370 |
| PFS* | |
| Médiane en mois (IC 95 %) | 2,2 (2,1 - 3,4) |
| PFS à 6 mois | 33 % |
| PFS à 12 mois | 22 % |
| OS | |
| Médiane en mois (IC 95 %) | 11,3 (9,7 -13,1) |
| OS à 6 mois | 67 % |
| OS à 12 mois | 47 % |
† Sur la base de la meilleure réponse de la maladie, stable ou meilleur
‡ Sur la base des estimations de Kaplan-Meier ; inclut 84 patients avec une réponse de 6 mois ou plus
Une analyse a été réalisée dans l'étude KEYNOTE-052 chez les patients qui avaient des tumeurs exprimant PD-L1 avec un score positif combiné (CPS) < 10 (n = 251 ; 68 %) ou ≥ 10 (n = 110 ; 30 %) sur la base du kit PD-L1 IHC 22C3 pharmDxTM (voir tableau 24).
Tableau 24 : ORR et OS selon l'expression de PD-L1
| Critère | CPS < 10n = 251 | CPS ≥ 10n = 110 |
| Taux de réponse objective* | ||
| ORR %, (IC 95 %) | 20 % (16 - 26) | 47 % (38 - 57) |
| OS | ||
| Médiane en mois (IC 95 %) | 10 (8 - 12) | 19 (12 - 29) |
| OS à 12 mois | 41 % | 61 % |
* En aveugle par une revue centralisée indépendante utilisant RECIST 1.1
KEYNOTE-361 est une étude clinique de phase III, ouverte, randomisée, contrôlée évaluant le pembrolizumab associé ou non à une chimiothérapie à base de sels de platine (c'est-à-dire le cisplatine ou le carboplatine avec la gemcitabine) versus chimiothérapie, en traitement de première ligne chez des sujets atteints de carcinome urothélial avancé ou métastatique. Les résultats de l'étude KEYNOTE-361 pour le pembrolizumab en association avec une chimiothérapie n'ont pas montré d'amélioration statistiquement significative de la PFS telle qu'évaluée en aveugle par une revue centralisée indépendante utilisant RECIST 1.1 (HR 0,78 ; IC 95 % : 0,65 - 0,93 ; p = 0,0033), ni de l'OS (HR 0,86 ; IC 95 % : 0,72 - 1,02 ; p = 0,0407) versus la chimiothérapie seule. Selon l'ordre prédéfini des tests hiérarchisés, aucun test formel de significativité statistique du pembrolizumab versus chimiothérapie n'a pu être effectué. Les principaux résultats d'efficacité du pembrolizumab en monothérapie chez les patients pour lesquels le carboplatine a été sélectionné par l'investigateur plutôt que le cisplatine comme le meilleur choix de chimiothérapie étaient cohérents avec les résultats de l'étude KEYNOTE-052. Les résultats d'efficacité chez les patients dont les tumeurs expriment PD-L1 avec un CPS ≥ 10 étaient similaires à la population globale pour laquelle le carboplatine a été sélectionné comme la chimiothérapie de choix. Voir le tableau 25 et les figures 18 et 19.
Tableau 25 : Réponse à pembrolizumab 200 mg toutes les 3 semaines ou à la chimiothérapie chez les patients atteints d'un carcinome urothélial non traité précédemment pour lesquels le carboplatine a été sélectionné par l'investigateur plutôt que le cisplatine comme le meilleur choix de chimiothérapie dans l'étude KEYNOTE-361
| Critèred'évaluation | Pembrolizumabn = 170 | Chimiothérapien = 196 | Pembrolizumab CPS ≥ 10n = 84 | Chimiothérapie CPS ≥ 10n = 89 |
| Taux de réponse objective* | ||||
| ORR %, (IC 95 %) | 28 % (21,1 - 35,0) | 42 % (34,8 - 49,1) | 30 % (20,3 - 40,7) | 46 % (35,4 - 57,0) |
| Réponse complète | 10 % | 11 % | 12 % | 18 % |
| Réponse partielle | 18 % | 31 % | 18 % | 28 % |
| Durée de réponse* | ||||
| Médiane en mois (intervalle) | NR (3,2+ - 36,1+) | 6,3 (1,8+ - 33,8+) | NR (4,2 - 36,1+) | 8,3 (2,1+ - 33,8+) |
| % avecdurée ≥ 12 mois† | 57 % | 30 % | 63 % | 38 % |
| PFS* | ||||
| Médiane en mois (IC 95 %) | 3,2 (2,2 - 5,5) | 6,7 (6,2 - 8,1) | 3,9 (2,2 - 6,8) | 7,9 (6,1 - 9,3) |
| PFS à 12 mois | 25 % | 24 % | 26 % | 31 % |
| OS | ||||
| Médiane en mois (IC 95 %) | 14,6 (10,2 - 17,9) | 12,3 (10,0 - 15,5) | 15,6 (8,6 - 19,7) | 13,5 (9,5 - 21,0) |
| OS à 12 mois | 54 % | 51 % | 57 % | 54 % |
† Sur la base des estimations de Kaplan-Meier

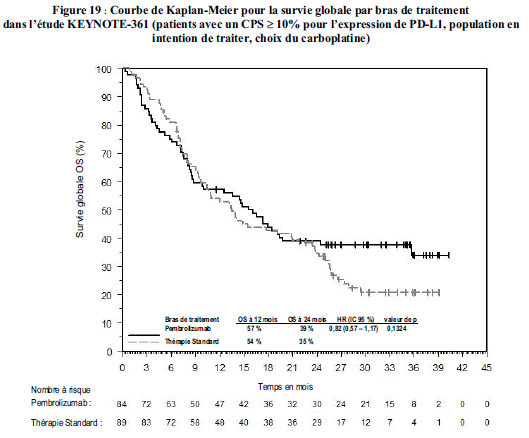
Carcinome épidermoïde de la tête et du cou (CETEC)
KEYNOTE-048 : Etude contrôlée du traitement en monothérapie et en association chez des patients atteints d'un CETEC naïfs de traitement à un stade métastatique ou en récidive
L'efficacité de pembrolizumab a été étudiée dans KEYNOTE-048, une étude multicentrique, randomisée, en ouvert, contrôlée par traitement actif, chez des patients atteints d'un CETEC histologiquement confirmé récidivant ou métastatique de la cavité orale, du pharynx ou du larynx qui n'avaient pas reçu préalablement de traitement systémique pour une maladie récidivante ou métastatique et qui étaient considérés non éligibles aux traitements locaux. Les patients atteints d'un carcinome nasopharyngé, d'une maladie auto-immune active ayant nécessité un traitement systémique dans les 2 ans précédents, ou dont l'état clinique nécessitait un traitement immunosuppresseur, étaient inéligibles pour l'étude. La randomisation était stratifiée selon l'expression de PD-L1 (TPS ≥ 50% ou <50 %), le statut HPV (positif ou négatif) et le statut de performance ECOG (0 vs 1). Les patients étaient randomisés 1:1:1 dans l'un des bras de traitement suivants :
· Pembrolizumab 200 mg toutes les 3 semaines· Pembrolizumab 200 mg toutes les 3 semaines, carboplatine ASC 5 mg/mL/min toutes les 3 semaines ou cisplatine 100 mg/m2 toutes les 3 semaines et 5-FU 1 000 mg/m2/j pendant 4 jours en continu toutes les 3 semaines (avec un maximum de 6 cycles de platine et 5-FU).
· Cétuximab à une dose de charge de 400 mg/m2 puis 250 mg/m2 une fois par semaine, carboplatine ASC 5 mg/mL/min toutes les 3 semaines ou cisplatine 100 mg/m2 toutes les 3 semaines et 5-FU 1 000 mg/m2/j pendant 4 jours en continu toutes les 3 semaines (avec un maximum de 6 cycles de platine et 5-FU).
Le traitement par pembrolizumab était poursuivi jusqu'à progression de la maladie définie par RECIST 1.1 telle que déterminée par l'investigateur, toxicité inacceptable ou durée maximale de 24 mois.
L'administration de pembrolizumab était autorisée au-delà de la progression de la maladie définie par RECIST si le patient était cliniquement stable et si l'investigateur considérait qu'il y avait un bénéfice clinique. Une évaluation de la réponse tumorale était réalisée à la semaine 9 et ensuite toutes les 6 semaines pendant la première année, puis toutes les 9 semaines jusqu'à 24 mois.
Parmi les 882 patients de KEYNOTE-048, 754 (85 %) avaient des tumeurs qui exprimaient PD-L1 avec un CPS ≥ 1 basé sur le kit PD-L1 IHC 22C3 pharmDxTM. Les caractéristiques de ces 754 patients à l'inclusion incluaient : âge médian de 61 ans (de 20 à 94 ans) ; 36 % âgés de 65 ans ou plus ; 82 % d'hommes ; 74 % de type caucasien et 19 % de type asiatique ; le statut de performance ECOG était de 1 chez 61 % ; et 77 % étaient d'anciens fumeurs ou des fumeurs actuels. Les caractéristiques de la maladie étaient : 21 % de tumeurs HPV positives et 95 % présentaient une maladie au stade IV (stade IVa : 21 %, stade IVb : 6 % et stade IVc : 69 %).
Les principaux critères d'ef icacité étaient l'OS et la PFS (évaluées en aveugle par une revue centralisée indépendante utilisant RECIST 1.1). L'étude a démontré une amélioration statistiquement significative de l'OS pour tous les patients randomisés dans le groupe pembrolizumab en association à la chimiothérapie par rapport au traitement standard (HR 0,72 ; IC 95 % 0,60-0,87), et pour les patients dont les tumeurs exprimaient PD-L1 avec un CPS ≥ 1 randomisés dans le groupe pembrolizumab en monothérapie par rapport au traitement standard. Les tableaux 26 et 27 résument les résultats clés des critères d'efficacité pour le pembrolizumab chez les patients dont les tumeurs expriment PD-L1 avec un CPS ≥ 1 dans l'étude KEYNOTE-048 lors de l'analyse finale réalisée à un suivi médian de 13 mois pour le pembrolizumab en association à la chimiothérapie et à un suivi médian de 11,5 mois pour le pembrolizumab en monothérapie. Les figures 20 et 21 montrent les courbes de Kaplan-Meier pour l'OS basées sur l'analyse finale.
Tableau 26 : Résultats d'ef icacité pour pembrolizumab plus chimiothérapie dans KEYNOTE-048 avec expression de PD-L1 (CPS ≥ 1)
| Critère d'évaluation | Pembrolizumab + Chimiothérapie à base de sels de platine + 5-FUn = 242 | Traitement standard* n = 235 |
| OS | ||
| Nombre (%) de patients avec événement | 177 (73 %) | 213 (91 %) |
| Médiane en mois (IC 95 %) | 13,6 (10,7 - 15,5) | 10,4 (9,1 - 11,7) |
| Hazard ratio† (IC 95 %) | 0,65 (0,53 - 0,80) | |
| Valeur de p‡ | 0,00002 | |
| PFS | ||
| Nombre (%) de patients avec événement | 212 (88 %) | 221 (94 %) |
| Médiane en mois (IC 95 %) | 5,1 (4,7 - 6,2) | 5,0 (4,8 - 6,0) |
| Hazard ratio† (IC 95 %) | 0,84 (0,69 - 1,02) | |
| Valeur de p‡ | 0,03697 | |
| Taux de réponse objective | ||
| ORR§ % (IC 95 %) | 36 % (30,3 - 42,8) | 36 % (29,6 - 42,2) |
| Réponse complète | 7 % | 3 % |
| Réponse partielle | 30 % | 33 % |
| Valeur de p¶ | 0,4586 | |
| Durée de réponse | ||
| Médiane en mois (intervalle) | 6,7 (1,6+ - 39,0+) | 4,3 (1,2+ - 31,5+) |
| % avec durée ≥ 6 mois | 54 % | 34 % |
† Sur la base du modèle « Cox proportional hazard » stratifié
‡ Sur la base du test de log rank stratifié
§ Réponse : meilleure réponse objective telle que réponse complète ou partielle confirmées
¶ Sur la base de la méthode de Miettinen et Nurminen stratifiée par ECOG (0 vs. 1), statut HPV (positifvs. négatif) et statut PD-L1 (fortement positif vs. non fortement positif)
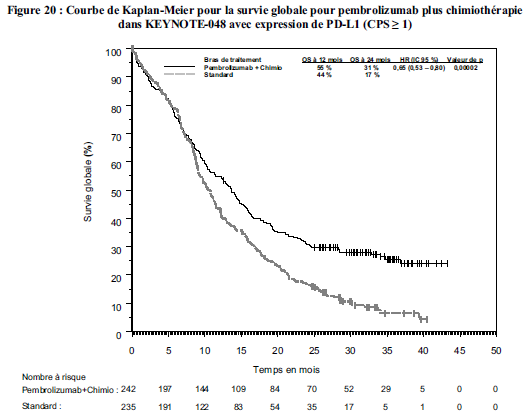
Tableau 27 : Résultats d'ef icacité pour pembrolizumab en monothérapie dans KEYNOTE-048 avec expression de PD-L1 (CPS ≥1)
| Critère d'évaluation | Pembrolizumab n = 257 | Traitement standard* n = 255 |
| OS | ||
| Nombre (%) de patients avecévénement | 197 (77 %) | 229 (90 %) |
| Médiane en mois (IC 95 %) | 12,3 (10,8 - 14,3) | 10,3 (9,0 - 11,5) |
| Hazard ratio† (IC 95 %) | 0,74 (0,61 - 0,90) | |
| Valeur de p‡ | 0,00133 | |
| PFS | ||
| Nombre (%) de patients avec événement | 228 (89 %) | 237 (93 %) |
| Médiane en mois (IC 95 %) | 3,2 (2,2 - 3,4) | 5,0 (4,8 - 6,0) |
| Hazard ratio† (IC 95 %) | 1,13 (0,94 - 1,36) | |
| Valeur de p‡ | 0,89580 | |
| Taux de réponse objective | ||
| ORR§ % (IC 95 %) | 19,1 % (14,5 - 24,4) | 35 % (29,1 - 41,1) |
| Réponse complète | 5 % | 3 % |
| Réponse partielle | 14 % | 32 % |
| Valeur de p¶ | 1,0000 | |
| Durée de réponse | ||
| Médiane en mois (intervalle) | 23,4 (1,5+ - 43,0+) | 4,5 (1,2+ - 38,7+) |
| % avec durée ≥ 6 mois | 81 % | 36 % |
† Sur la base du modèle « Cox proportional hazard » stratifié
‡ Sur la base du test de log rank stratifié
§ Réponse : meilleure réponse objective telle que réponse complète ou partielle confirmées
¶ Sur la base de la méthode de Miettinen et Nurminen stratifiée par ECOG (0 vs. 1), statut HPV (positif vs. négatif) et statut PD-L1 (fortement positif vs. non fortement positif)
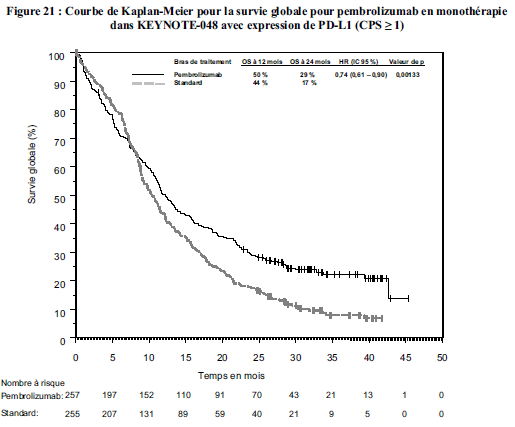
Une analyse a été réalisée dans KEYNOTE-048 chez les patients dont les tumeurs exprimaient PD-L1 avec CPS ≥ 20 [pembrolizumab plus chimiothérapie : n = 126 (49 %) vs. traitement standard : n = 110 (43 %) et pembrolizumab en monothérapie : n = 133 (52 %) vs. traitement standard : n = 122 (48 %)] (voir Tableau 28).
Tableau 28 : Résultat d'ef icacité pour pembrolizumab plus chimiothérapie et pembrolizumab en monothérapie selon l'expression de PD-L1 dans KEYNOTE 048 (CPS ≥ 20)
| Endpoint | Pembrolizumab + Chimiothérapie platine + 5-FUn = 126 | Traitementstandard* n = 110 | Pembrolizumab en monothérapie n = 133 | Traitementstandard* n = 122 |
| OS | ||||
| Nombre (%) de patients avec événement | 84 (66,7 %) | 98 (89,1 %) | 94 (70,7 %) | 108 (88,5 %) |
| Médiane en mois (IC 95 %) | 14,7 (10,3 - 19,3) | 11,0 (9,2 - 13,0) | 14,8 (11,5 - 20,6) | 10,7 (8,8 - 12,8) |
| Hazard ratio† (IC 95 %) | 0,60 (0,45 - 0,82) | 0,58 (0,44 - 0,78) | ||
| Valeur de p‡ | 0,00044 | 0,00010 | ||
| OS à 6 mois (IC 95 %) | 74,6 (66,0 - 81,3) | 80,0 (71,2 - 86,3) | 74,4 (66,1 - 81,0) | 79,5 (71,2 - 85,7) |
| OS à 12 mois (IC 95 %) | 57,1 (48,0 - 65,2) | 46,1 (36,6 - 55,1) | 56,4 (47,5 - 64,3) | 44,9 (35,9 - 53,4) |
| OS à 24 mois (IC 95 %) | 35,4 (27,2 - 43,8) | 19,4 (12,6 - 27,3) | 35,3 (27,3 - 43,4) | 19,1 (12,7 - 26,6) |
| PFS | ||||
| Nombre (%) de patients avec événement | 106 (84,1 %) | 104 (94,5 %) | 115 (86,5 %) | 114 (93,4 %) |
| Médiane en mois (IC 95 %) | 5,8 (4,7 - 7,6) | 5,3 (4,9 - 6,3) | 3,4 (3,2 - 3,8) | 5,3 (4,8 - 6,3) |
| Hazard ratio† (IC 95 %) | 0,76 (0,58 - 1,01) | 0,99 (0,76 - 1,29) | ||
| Valeur de p‡ | 0,02951 | 0,46791 | ||
| PFS à 6 mois (IC 95 %) | 49,4 (40,3 - 57,9) | 47,2 (37,5 - 56,2) | 33,0 (25,2 - 41,0) | 46,6 (37,5 - 55,2) |
| PFS à 12 mois (IC 95 %) | 23,9 (16,7 - 31,7) | 14,0 (8,2 - 21,3) | 23,5 (16,6 - 31,1) | 15,1 (9,3 - 22,2) |
| PFS à 24 mois (IC 95 %) | 14,6 (8,9 - 21,5) | 5,0 (1,9 - 10,5) | 16,8 (10,9 - 23,8) | 6,1 (2,7 - 11,6) |
| Taux de réponse objective | ||||
| ORR§ % (IC 95 %) | 42,9 (34,1 - 52,0) | 38,2 (29,1 - 47,9) | 23,3 (16,4 - 31,4) | 36,1 (27,6 - 45,3) |
| Durée de réponse | ||||
| Nombre de répondeurs | 54 | 42 | 31 | 44 |
| Médiane en mois (intervalle) | 7,1 (2,1+ - 39,0+) | 4,2 (1,2+ - 31,5+) | 22,6 (2,7+ - 43,0+) | 4,2 (1,2+ - 31,5+) |
† Sur la base du modèle « Cox proportional hazard » stratifié
‡ Sur la base du test de log rank stratifié
§ Réponse : meilleure réponse objective telle que réponse complète ou partielle confirmées
Une analyse exploratoire de sous-groupes a été réalisée dans KEYNOTE-048 chez des patients dont les tumeurs exprimaient PD-L1 avec 1 ≤ CPS < 20 [pembrolizumab plus chimiothérapie : n = 116 (45 %) vs. traitement standard : n = 125 (49 %) et pembrolizumab en monothérapie : n = 124 (48 %) vs. traitement standard : n = 133 (52 %)] (voir Tableau 29).
Tableau 29 : Résultats d'ef icacité de pembrolizumab plus chimiothérapie et pembrolizumab en monothérapie selon l'expression de PD-L1 dans KEYNOTE 048 (1 ≤ CPS < 20)
| Critère d'évaluation | Pembrolizumab + Chimiothérapie à base de sels de platine + 5-FUn = 116 | Traitementstandard* n = 125 | Pembrolizumab en monothérapie n = 124 | Traitementstandard* n = 133 |
| OS | ||||
| Nombre (%) de patients avecévénement | 93 (80,2 %) | 115 (92,0 %) | 103 (83,1 %) | 121 (91,0 %) |
| Médiane en mois (IC 95 %) | 12,7 (9,4 - 15,3) | 9,9 (8,6 - 11,5) | 10,8 (9,0 - 12,6) | 10,1 (8,7 - 12,1) |
| Hazard ratio† (IC 95 %) | 0,71 (0,54 - 0,94) | 0,86 (0,66 - 1,12) | ||
| OS à 6 mois (IC 95 %) | 76,7 (67,9 - 83,4) | 77,4 (69,0 - 83,8) | 67,6 (58,6 - 75,1) | 78,0 (70,0 - 84,2) |
| OS à 12 mois (IC 95 %) | 52,6 (43,1 - 61,2) | 41,1 (32,4 - 49,6) | 44,0 (35,1 - 52,5) | 42,4 (33,9 - 50,7) |
| OS à 24 mois (IC 95 %) | 25,9 (18,3 - 34,1) | 14,5 (9,0 - 21,3) | 22,0 (15,1 - 29,6) | 15,9 (10,3 - 22,6) |
| PFS | ||||
| Nombre (%) de patients avec événement | 106 (91,4 %) | 117 (93,6 %) | 113 (91,1 %) | 123 (92,5 %) |
| Médiane en mois (IC 95 %) | 4,9 (4,2 - 5,3) | 4,9 (3,7 - 6,0) | 2,2 (2,1 - 2,9) | 4,9 (3,8 - 6,0) |
| Hazard ratio† (IC 95 %) | 0,93 (0,71 - 1,21) | 1,25 (0,96 - 1,61) | ||
| PFS à 6 mois (IC 95 %) | 40,1 (31,0 - 49,0) | 40,0 (31,2 - 48,5) | 24,2 (17,1 - 32,0) | 41,4 (32,8 - 49,7) |
| PFS à 12 mois (IC 95 %) | 15,1 (9,1 - 22,4) | 11,3 (6,4 - 17,7) | 17,5 (11,4 - 24,7) | 12,1 (7,2 - 18,5) |
| PFS à 24 mois (IC 95 %) | 8,5 (4,2 - 14,7) | 5,0 (1,9 - 10,1) | 8,3 (4,3 - 14,1) | 6,3 (2,9 - 11,5) |
| Taux de réponse objective | ||||
| ORR‡ % (IC 95 %) | 29,3 (21,2 - 38,5) | 33,6 (25,4 - 42,6) | 14,5 (8,8 - 22,0) | 33,8 (25,9 - 42,5) |
| Durée de réponse | ||||
| Nombre de répondeurs | 34 | 42 | 18 | 45 |
| Médiane en mois (intervalle) | 5,6 (1,6+ - 25,6+) | 4,6 (1,4+ - 31,4+) | NA (1,5+ - 38,9+) | 5,0 (1,4+ - 38,7+) |
† Sur la base du modèle « Cox proportional hazard » stratifié
‡ Réponse : meilleure réponse objective telle que réponse complète ou partielle confirmées
KEYNOTE-040 : Etude contrôlée chez des patients atteints d'un CETEC précédemment traités par une chimiothérapie à base de sels de platine
L'efficacité et la tolérance de pembrolizumab ont été étudiées dans KEYNOTE-040, une étude multicentrique, randomisée, en ouvert, contrôlée, dans le traitement du CETEC histologiquement confirmé de la cavité orale, du pharynx ou du larynx récidivant ou métastatique chez des patients qui présentaient une progression de la maladie pendant ou après une chimiothérapie à base de sels de platine administrée pour un CETEC récidivant ou métastatique ou après une chimiothérapie à base de sels de platine administrée dans le cadre d'un traitement d'induction, concomitant ou adjuvant, et qui n'étaient pas candidats à une thérapie locale à visée curative. Les patients ont été stratifiés selon l'expression de PD-L1 (TPS ≥ 50%), le statut HPV et le statut de performance ECOG, et ensuite randomisés (1:1) pour recevoir soit du pembrolizumab 200 mg toutes les 3 semaines (n = 247) soit un des trois traitements standards (n = 248) : méthotrexate 40 mg/m2 une fois par semaine (n = 64), docétaxel 75 mg/m2 une fois toutes les 3 semaines (n = 99) ou cétuximab à une dose de charge de 400 mg/m2 puis 250 mg/m2 une fois par semaine (n = 71). Le traitement pouvait être poursuivi au-delà de la progression de la maladie si le patient était cliniquement stable et si l'investigateur considérait qu'il y avait un bénéfice clinique. L'étude excluait les patients atteints d'un carcinome nasopharyngé, d'une maladie auto-immune active ayant nécessité un traitement systémique dans les 2 ans précédents, les patients dont l'état clinique nécessitait un traitement immunosuppresseur ou ceux qui avaient précédemment reçus 3 traitements ou plus pour un CETEC récidivant et/ou métastatique. Une évaluation de la réponse tumorale était réalisée à 9 semaines, puis toutes les 6 semaines jusqu'à la Semaine 52, et ensuite toutes les 9 semaines jusqu'à 24 mois.
Parmi les 495 patients de KEYNOTE-040, 129 (26 %) avaient des tumeurs qui exprimaient PD-L1 avec un TPS ≥ 50 % avec le kit PD-L1 IHC 22C3 pharmDxTM. Les caractéristiques à l'inclusion de ces 129 patients comprenaient : âge médian de 62 ans (40 % âgés de 65 ans ou plus) ; 81 % d'hommes ; 78 % de type caucasien, 11 % de type asiatique et 2 % de type noir ; le statut de performance ECOG était de 0 ou 1 chez 23 % et 77 % des patients, respectivement ; et 19 % présentaient des tumeurs HPV positive. Soixante-sept pourcents (67 %) des patients présentaient une maladie au stade M1 et la majorité au stade IV (stade IV : 32 %, stade IVa : 14 %, stade IVb : 4 % et stade IVc : 44 %). Seize pourcents (16 %) présentaient une progression de la maladie après une chimiothérapie néoadjuvante ou adjuvante à base de sels de platine et 84 % avaient reçu 1-2 traitements systémiques antérieurs pour la maladie métastatique.
Le critère principal d'évaluation de l'ef icacité dans la population en intention de traiter (ITT) était l'OS. L'analyse initiale a abouti à un HR pour l'OS de 0,82 (IC 95 % : 0,67 - 1,01) avec une valeur unilatérale de p de 0,0316. L'OS médiane était de 8,4 mois pour le pembrolizumab par rapport à 7,1 mois pour le traitement standard. Le tableau 30 résume les critères principaux d'ef icacité pour la population avec un TPS ≥ 50 %. La courbe de Kaplan-Meier pour l'OS pour la population avec un TPS ≥ 50 % est représentée en Figure 22.
Tableau 30 : Efficacité du pembrolizumab 200 mg toutes les 3 semaines chez les patients atteints de CETEC avec un TPS ≥ 50 % précédemment traités par une chimiothérapie à base de sels de platine dans KEYNOTE-040
| Critère | Pembrolizumab 200 mg toutes les 3 semainesn = 64 | Traitement standard* n = 65 |
| OS | ||
| Nombre (%) de patients avec événement | 41 (64 %) | 56 (86 %) |
| Hazard ratio† (IC 95 %) | 0,53 (0,35 - 0,81) | |
| Valeur de p ‡ | 0,001 | |
| Médiane en mois (IC 95 %) | 11,6 (8,3 - 19,5) | 6,6 (4,8 - 9,2) |
| PFS§ | ||
| Nombre (%) de patients avec événement | 52 (81 %) | 58 (89 %) |
| Hazard ratio† (IC 95 %) | 0,58 (0,39 - 0,86) | |
| Valeur de p ‡ | 0,003 | |
| Médiane en mois (IC 95 %) | 3,5 (2,1 - 6,3) | 2,1 (2,0 - 2,4) |
| Taux (%) à 6 mois (IC 95 %) | 40,1 (28,1 - 51,9) | 17,1 (8,8 - 27,7) |
| Taux de réponse objective§ | ||
| ORR % (IC 95 %) | 26,6 (16,3 - 39,1) | 9,2 (3,5 - 19,0) |
| Valeur de p ¶ | 0,0009 | |
| Réponse complète | 5 % | 2 % |
| Réponse partielle | 22 % | 8 % |
| Maladie stable | 23 % | 23 % |
| Durée de réponse §,# | ||
| Médiane en mois (intervalle) | Non atteint (2,7 - 13,8+) | 6,9 (4,2 - 18,8) |
| Nombre (%Þ) de patients avec durée≥ 6 mois | 9 (66) | 2 (50) |
† Hazard ratio (pembrolizumab comparé au traitement standard) sur la base du modèle « Cox proportional hazard » stratifié
‡ Valeur de p unilatérale sur la base du test de Log rank
§ Evalué en aveugle par une revue centralisée indépendante utilisant RECIST 1.1
¶ Sur la base de la méthode de Miettinen et Nurminen
# Sur la base des patients ayant une meilleure réponse objective telle que réponse complète ou partielle confirmées
Þ Sur la base de l'estimation de Kaplan Meier
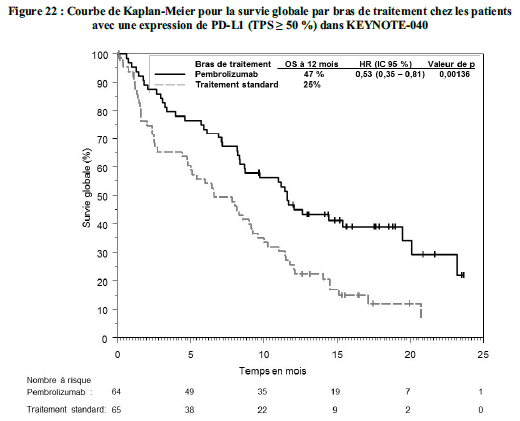
Carcinome à cellules rénales
KEYNOTE-426 : Etude contrôlée du traitement en association à l'axitinib chez les patients atteints d'un CCR naïfs de traitement
L'efficacité de pembrolizumab en association à l'axitinib a été étudiée dans KEYNOTE-426, une étude randomisée, multicentrique, en ouvert, contrôlée par traitement actif, réalisée chez des patients atteints d'un CCR avancé avec une composante à cellules claires, quel que soit le statut d'expression tumorale de PD-L1 et les groupes de risque de l'International Metastatic RCC Database Consortium (IMDC). L'étude excluait les patients atteints d'une maladie auto-immune ou ceux dont l'état clinique nécessitait un traitement immunosuppresseur. La randomisation était stratifiée selon les groupes de risque (favorable versus intermédiaire versus défavorable) et la région géographique (Amérique du Nord versus Europe de l'Ouest versus « reste du monde »). Les patients étaient randomisés (1:1) dans l'un des bras de traitement suivants :
· pembrolizumab 200 mg toutes les 3 semaines par voie intraveineuse en association à l'axitinib 5 mg par voie orale, deux fois par jour. Une augmentation de la dose d'axitinib à 7 mg deux fois par jour était autorisée chez les patients qui toléraient l'axitinib 5 mg deux fois par jour pendant 2 cycles consécutifs de traitement (c'est-à-dire 6 semaines) sans aucun effet indésirable > Grade 2 lié à l'axitinib et avec une pression artérielle bien contrôlée à ≤ 150/90 mm Hg. Une augmentation de la dose d'axitinib à 10 mg deux fois par jour était autorisée en utilisant les mêmes critères. L'axitinib pouvait être arrêté ou réduit à 3 mg deux fois par jour puis 2 mg deux fois par jour pour prendre en charge la toxicité.· sunitinib 50 mg par voie orale, une fois par jour pendant 4 semaines, suivi de 2 semaines de fenêtre thérapeutique.
Le traitement par pembrolizumab et axitinib a été poursuivi jusqu'à progression de la maladie définie par RECIST v1.1 telle que déterminée en aveugle par une revue centralisée indépendante ou confirmée par l'investigateur, toxicité inacceptable, ou bien pour pembrolizumab une durée maximale de 24 mois.
L'administration de pembrolizumab et axitinib était autorisée au-delà de la progression de la maladie définie par RECIST si le patient était cliniquement stable et si l'investigateur considérait qu'il y avait un bénéfice clinique. L'évaluation tumorale était réalisée à l'inclusion, après randomisation à la semaine 12 puis toutes les 6 semaines jusqu'à la semaine 54 et ensuite toutes les 12 semaines.
Un total de 861 patients a été randomisé. Les caractéristiques de la population de l'étude étaient : âge médian de 62 ans (de 26 à 90 ans) ; 38 % âgés de 65 ans ou plus ; 73 % d'hommes ; 79 % de type caucasien et 16 % de type asiatique ; 80 % avaient un Score de Performance Karnofsky (SPK) de 90-100 et 20 % avaient un SPK de 70-80 ; la répartition des patients par groupe de risque IMDC était : 31 % favorable, 56 % intermédiaire et 13 % défavorable.
Les critères principaux d'évaluation de l'efficacité étaient l'OS et la PFS (tels qu'évalués en aveugle par une revue centralisée indépendante utilisant RECIST 1.1). Les critères secondaires d'évaluation de l'efficacité étaient l'ORR et la durée de réponse, tel qu'évalués en aveugle par une revue centralisée indépendante utilisant RECIST 1.1. L'étude a démontré une amélioration statistiquement significative de l'OS (HR 0,53 ; IC 95 % : 0,38 - 0,74 ; valeur de p = 0,00005) et de la PFS (HR 0,69 ; IC 95 % : 0,56 - 0,84 ; valeur de p = 0,00012) chez les patients randomisés dans le bras pembrolizumab en association par rapport au sunitinib lors de son analyse intermédiaire prédéfinie. Le tableau 31 résume les critères clés d'efficacité et les figures 23 et 24 représentent les courbes de Kaplan-Meier pour l'OS et la PFS, basées sur l'analyse finale avec un suivi médian de 37,7 mois.
Tableau 31 : Résultats d'ef icacité dans l'étude KEYNOTE-426
| Critère d'évaluation | Pembrolizumab Axitinibn = 432 | Sunitinib n = 429 |
| OS | ||
| Nombre (%) de patients avec événement | 193 (45 %) | 225 (52 %) |
| Médiane en mois (IC 95 %) | 45,7 (43,6 - ND) | 40,1 (34,3 - 44,2) |
| Hazard ratio* (IC 95 %) | 0,73 (0,60 - 0,88) | |
| Valeur de p† | 0,00062 | |
| PFS‡ | ||
| Nombre (%) de patients avec événement | 286 (66 %) | 301 (70 %) |
| Médiane en mois (IC 95 %) | 15,7 (13,6 - 20,2) | 11,1 (8,9 - 12,5) |
| Hazard ratio* (IC 95 %) | 0,68 (0,58 - 0,80) | |
| Valeur de p† | < 0,00001 | |
| Taux de réponse objective | ||
| ORR§ % (IC 95%) | 60 (56 - 65) | 40 (35 - 44) |
| Réponse complète | 10 % | 3 % |
| Réponse partielle | 50 % | 36 % |
| Valeur de p¶ | < 0,0001 | |
| Durée de réponse | ||
| Médiane en mois (intervalle) | 23,6 (1,4+ - 43,4+) | 15,3 (2,3 - 42,8+) |
| Nombre (%#) de patients avecdurée ≥ 30 mois | 87 (45 %) | 29 (32 %) |
† Valeur de p nominale sur la base du test de Log rank stratifié
‡ Evalué en aveugle par une revue centralisée indépendante utilisant RECIST 1.1
§ Sur la base des patients ayant une meilleure réponse objective telle que réponse complète ou partielle confirmées
¶ Valeur de p nominale sur la base de la méthode de Miettinen et Nurminen stratifiée par groupe de risque IMDC et par région géographique. Lors de l'analyse intermédiaire prédéfinie de l'ORR (durée de suivi médiane de 12,8 mois), une supériorité statistiquement significative a été atteinte pour l'ORR en comparant pembrolizumab plus axitinib avec sunitinib Valeur de p < 0,0001.
# Sur la base de l'estimation de Kaplan-Meier
ND = non disponible
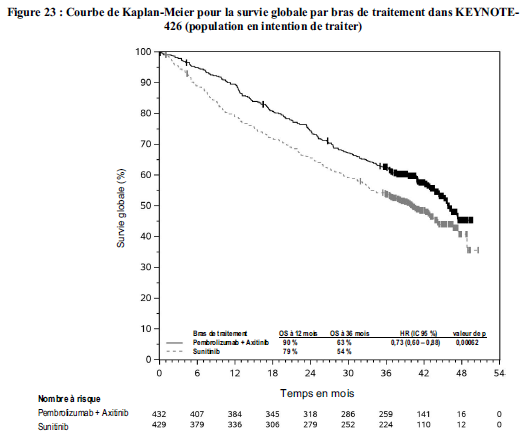
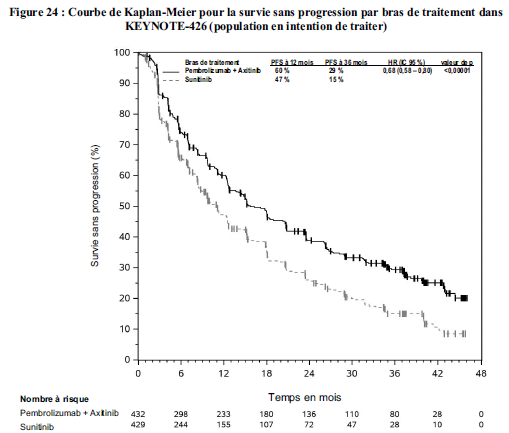
Des analyses en sous-groupes ont été réalisées dans KEYNOTE-426 chez les patients qui avaient une tumeur exprimant PD-L1 avec un score positif combiné (CPS) ≥ 1 [association pembrolizumab/axitinib : n = 243 (56 %) vs. sunitinib : n = 254 (59 %)] et CPS < 1 [association pembrolizumab/axitinib : n = 167 (39 %) vs. sunitinib : n = 158 (37 %)]. Des bénéfices en OS et PFS ont été observés quel que soit le niveau d'expression de PD-L1.
L'étude KEYNOTE-426 n'a pas été conçue pour évaluer l'ef icacité des sous-groupes individuels.
Lors de l'analyse intermédiaire prédéfinie pour les groupes de risque IMDC, le hazard ratio (HR) de l'OS pour les patients randomisés dans le bras pembrolizumab en association par rapport au sunitinib dans le groupe de risque favorable était de 0,64 (IC à 95 %, 0,24 - 1,68), pour le groupe de risque intermédiaire, le HR de l'OS était de 0,53 (IC à 95 %, 0,35 - 0,82) et pour le groupe de risque défavorable, le HR de l'OS était de 0,43 (IC à 95 %, 0,23 - 0,81). Les HR de la PFS (IC à 95 %) pour les groupes de risque favorable, intermédiaire et défavorable étaient de 0,81 (0,53 - 1,24), 0,69 (0,53 - 0,90) et 0,58 (0,35 - 0,94), respectivement. La différence d'ORR (IC à 95 %) pour les groupes de risque favorable, intermédiaire et défavorable était de 17,0 % (5,3 - 28,4), 25,5 % (16,7 - 33,9) et 31,5 % (15,7 - 46,2), respectivement.
Le tableau 32 résume les critères d'ef icacité selon les groupes de risque IMDC sur la base de l'analyse finale de l'OS à un suivi médian de 37,7 mois.
Tableau 32 : Résultats d'ef icacité dans l'étude KEYNOTE-426 par groupe de risque IMDC
| Critère* | Pembrolizumab +Axitinib n = 432 | Sunitinib n = 429 | Pembrolizumab + Axitinib vs. Sunitinib |
| OS | OS à 12 mois, % (IC 95 %) | OS HR (IC 95 %) | |
| Favorable | 95,6 (90,5 - 98,0) | 94,6 (89,0 - 97,4) | 1,17 (0,76 - 1,80) |
| Intermédiaire | 90,7 (86,2 - 93,8) | 77,6 (71,8 - 82,3) | 0,67 (0,52 - 0,86) |
| Défavorable | 69,6 (55,8 - 79,9) | 45,1 (31,2 - 58,0) | 0,51 (0,32 - 0,81) |
| PFS | Médiane (IC 95 %), mois | PFS HR (IC 95 %) | |
| Favorable | 20,7 (15,2 - 28,9) | 17,8 (12,5 - 20,7) | 0,76 (0,56 - 1,03) |
| Intermédiaire | 15,3 (12,5 - 20,8) | 9,7 (8,0 - 12,4) | 0,69 (0,55 - 0,86) |
| Défavorable | 4,9 (2,8 - 12,4) | 2,9 (2,7 - 4,2) | 0,53 (0,33 - 0,84) |
| ORR confirmé | % (IC 95 %) | Différence ORR,% (IC 95 %) | |
| Favorable | 68,8 (60,4 - 76,4) | 50,4 (41,5 - 59,2) | 18,5 (6,7 - 29,7) |
| Intermédiaire | 60,5 (54,0 - 66,8) | 39,8 (33,7 - 46,3) | 20,7 (11,8 - 29,2) |
| Défavorable | 39,3 (26,5 - 53,2) | 11,5 (4,4 - 23,4) | 27,7 (11,7 - 42,8) |
KEYNOTE-581 : Etude contrôlée du traitement en association au lenvatinib chez des patients atteints d'un CCR naïfs de traitement
L'efficacité de pembrolizumab en association au lenvatinib a été évaluée dans KEYNOTE-581, une étude multicentrique, en ouvert, randomisée conduite chez 1 069 patients atteints d'un CCR avancé avec un contingent à cellules claires comprenant d'autres composantes histologiques telles que sarcomatoïdes et papillaires, en traitement de première ligne. Les patients ont été inclus quel que soit le statut d'expression tumorale de PD-L1. L'étude excluait les patients atteints d'une maladie auto-immune active ou ceux dont l'état clinique nécessitait un traitement immunosuppresseur. La randomisation était stratifiée selon la région géographique (Amérique du Nord versus Europe de l'Ouest versus « reste du monde ») et les groupes pronostiques du Memorial Sloan Kettering Cancer Center (MSKCC) (favorable versus intermédiaire versus défavorable).
Les patients ont été randomisés (1 : 1 : 1) dans l'un des bras de traitement suivants :
· pembrolizumab 200 mg par voie intraveineuse toutes les 3 semaines jusqu'à 24 mois en association au lenvatinib 20 mg par voie orale, une fois par jour.· lenvatinib 18 mg par voie orale, une fois par jour en association à l'évérolimus 5 mg par voie orale, une fois par jour.
· sunitinib 50 mg par voie orale, une fois par jour pendant 4 semaines, suivi d'une fenêtre thérapeutique de 2 semaines.
Le traitement a été poursuivi jusqu'à toxicité inacceptable ou progression de la maladie telle que déterminée par l'investigateur et confirmée en aveugle par une revue centralisée indépendante utilisant RECIST 1.1.
L'administration de pembrolizumab avec le lenvatinib était autorisée au-delà de la progression de la maladie définie par RECIST si le patient était cliniquement stable et si l'investigateur considérait qu'il y avait un bénéfice clinique. Pembrolizumab a été poursuivi pendant 24 mois maximum ; cependant, le traitement par lenvatinib pouvait être poursuivi au-delà de 24 mois. Une évaluation du statut tumoral a été réalisée à l'inclusion, puis toutes les 8 semaines.
Parmi la population étudiée (355 patients dans le bras pembrolizumab associé au lenvatinib et 357 dans le bras sunitinib), les caractéristiques à l'inclusion étaient : âge médian de 62 ans (de 29 à 88 ans), 41 % âgés de 65 ans ou plus ; 74 % d'hommes ; 75 % de type Caucasien, 21 % de type Asiatique, 1 % de type Noir et 2 % d'autres origines ethniques ; 17 % et 83 % des patients avaient un score de Karnofsky à l'inclusion de 70 à 80 et 90 à 100, respectivement ; la répartition des patients par catégories de risque IMDC était de 33 % favorables, 56 % intermédiaires et 10 % défavorables, et par groupes pronostiques MSKCC était de 27 % favorables, 64 % intermédiaires et 9 % défavorables. La maladie métastatique était présente chez 99 % des patients et la maladie localement avancée était présente chez 1 %. Les sites les plus fréquents de métastases chez les patients étaient les poumons (69 %), les ganglions lymphatiques (46 %) et les os (26 %).
Le critère principal d'évaluation de l'efficacité était la PFS basée sur une évaluation en aveugle par une revue centralisée indépendante utilisant RECIST 1.1. Les critères secondaires clés d'évaluation de l'efficacité incluaient l'OS et l'ORR. L'étude a démontré des améliorations statistiquement significatives de la PFS, de l'OS et de l'ORR chez les patients randomisés dans le bras pembrolizumab en association au lenvatinib par rapport au bras sunitinib. La durée médiane de suivi de la survie était de 26,5 mois. La durée médiane de traitement par pembrolizumab plus lenvatinib était de 17,0 mois. Les résultats d'efficacité de KEYNOTE 581 sont résumés dans le tableau 33 et les figures 25 et 26. Les résultats de la PFS étaient cohérents entre les sous-groupes prédéfinis, les groupes pronostiques MSKCC et le statut d'expression tumorale de PD-L1. Les résultats d'efficacité selon le groupe pronostique MSKCC sont résumés dans le tableau 34.
Tableau 33 : Résultats d'ef icacité dans l'étude KEYNOTE-581
| Critère d'évaluation | Pembrolizumab 200 mg toutes les3 semaines et Lenvatinibn = 355 | Sunitinibn = 357 |
| PFS* | ||
| Nombre (%) de patients avec événement | 160 (45 %) | 205 (57 %) |
| Médiane en mois (IC 95 %) | 23,9 (20,8 - 27,7) | 9,2 (6,0 - 11,0) |
| Hazard ratio† (IC 95 %) | 0,39 (0,32 - 0,49) | |
| Valeur de p‡ | < 0,0001 | |
| OS | ||
| Nombre (%) de patients avec événement | 80 (23 %) | 101 (28 %) |
| Médiane en mois (IC 95 %) | NA (33,6 - NA) | NA (NA - NA) |
| Hazard ratio† (IC 95 %) | 0,66 (0,49 - 0,88) | |
| Valeur de p‡ | 0,0049 | |
| Taux de réponse objective | ||
| ORR§ % (IC 95%) | 71 % (66 - 76) | 36 % (31 - 41) |
| Réponse complète | 16 % | 4 % |
| Réponse partielle | 55 % | 32 % |
| Valeur de p¶ | < 0,0001 | |
| Durée de réponse# | ||
| Médiane en mois (intervalle) | 26 (1,6+ - 36,8+) | 15 (1,6+ - 33,2+) |
† Sur la base du modèle Cox proportional hazard stratifié
‡ Bilatérale basée sur le test de log rank stratifié
§ Réponse : meilleure réponse objective telle que réponse complète ou partielle confirmées
¶ Valeur de p nominale bilatérale basée sur le test de Cochran Mantel Haenszel (CMH) stratifié. Lors de l'analyse finale précédemment pré-définie de l'ORR (durée médiane de suivi de 17,3 mois), une supériorité statistiquement significative a été obtenue pour l'ORR comparant pembrolizumab plus lenvatinib versus sunitinib, (odds ratio : 3,84 [IC à 95 % : 2,81 - 5,26], valeur p < 0,0001).
# Sur la base des estimations de Kaplan-Meier
NA = non atteint
L'analyse primaire de l'OS n'a pas été ajustée pour tenir compte des traitements ultérieurs.
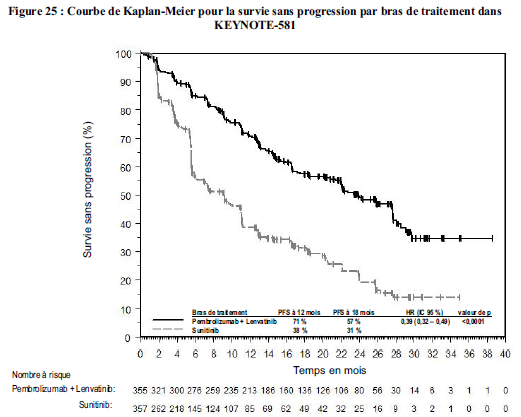
Une analyse actualisée de l'OS a été réalisée lorsque les patients recevant pembrolizumab et lenvatinib ou sunitinib avaient un suivi médian de la survie de 33,4 mois. Le hazard ratio était de 0,72 (IC à 95 % : 0,55 - 0,93) avec 105/355 (30 %) de décès dans le bras en association et 122/357 (34 %) de décès dans le bras sunitinib. Cette analyse actualisée de l'OS n'a pas été ajustée pour tenir compte des traitements ultérieurs.
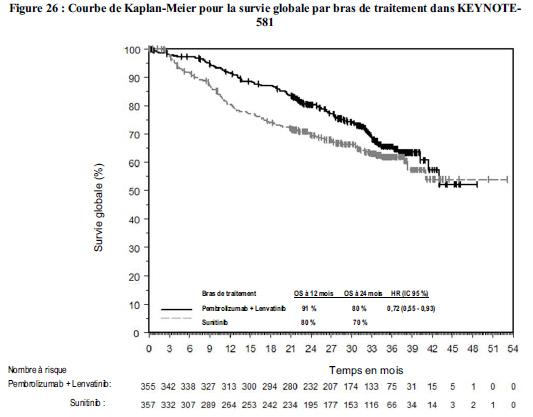
L'étude KEYNOTE-581 n'avait pas la puissance pour évaluer l'efficacité des sous-groupes individuels. Le tableau 34 résume les données d'efficacité selon le groupe pronostique MSKCC à partir de l'analyse primaire prédéfinie et de l'analyse de l'OS actualisée.
Tableau 34 : Résultats d'ef icacité dans l'étude KEYNOTE-581 par groupe pronostique MSKCC
| Pembrolizumab +Lenvatinib (n = 355) | Sunitinib (n = 357) | Pembrolizumab + Lenvatinib vs.Sunitinib | |||
| Nombrede patients | Nombred'évènements | Nombrede patients | Nombred'évènements | ||
| Survie sans progression (PFS) par une revue centralisée indépendante en aveugle* | PFS HR (IC 95 %) | ||||
| Favorable | 96 | 39 | 97 | 60 | 0,36 (0,23 - 0,54) |
| Intermédiaire | 227 | 101 | 228 | 126 | 0,44 (0,34 - 0,58) |
| Défavorable | 32 | 20 | 32 | 19 | 0,18 (0,08 - 0,42) |
| Survie globale (OS)* | OS HR (IC 95 %) | ||||
| Favorable† | 96 | 11 | 97 | 13 | 0,86 (0,38 - 1,92) |
| Intermédiaire | 227 | 57 | 228 | 73 | 0,66 (0,47 - 0,94) |
| Défavorable | 32 | 12 | 32 | 15 | 0,50 (0,23 - 1,08) |
| OS‡ actualisée | OS HR (IC 95 %) | ||||
| Favorable† | 96 | 17 | 97 | 17 | 1,00 (0,51 - 1,96) |
| Intermédiaire | 227 | 74 | 228 | 87 | 0,71 (0,52 - 0,97) |
| Défavorable | 32 | 14 | 32 | 18 | 0,50 (0,25 - 1,02) |
† L'interprétation des HR est limitée par le faible nombre d'événements (24/193 et 34/193)
‡ Suivi médian : 33,4 mois (cut-offdes données - 31 mars 2021)
KEYNOTE-564 : Etude contrôlée versus placebo pour le traitement adjuvant des patients avec un carcinome à cellules rénales (CCR) réséqué
L'efficacité de pembrolizumab a été évaluée en tant que traitement adjuvant d'un CCR dans KEYNOTE-564, une étude multicentrique, randomisée, en double-aveugle, contrôlée versus placebo chez 994 patients présentant un risque accru de récidive défini comme un risque intermédiaire-élevé ou élevé, ou qui présentaient un stade M1 sans signe de maladie (NED). La catégorie de risque intermédiaire-élevé comprenait : pT2 de Grade 4 ou avec des composantes sarcomatoïdes ; pT3 de tout Grade sans atteinte ganglionnaire (N0) ou métastases à distance (M0). La catégorie à haut risque comprenait : pT4 de tout Grade N0 et M0 ; tout pT et tout Grade avec atteinte ganglionnaire et M0. La catégorie M1 NED comprenait des patients atteints d'une maladie métastatique qui avaient subi une résection complète des lésions primaires et métastatiques. Les patients devaient avoir subi une néphrectomie partielle ou une néphrectomie totale (et une résection totale des lésions métastatiques des tissus mous, solides et isolées chez les participants M1 NED) avec des marges chirurgicales négatives ≥ 4 semaines avant la sélection. L'étude excluait les patients atteints d'une maladie auto-immune active ou dont l'état clinique nécessitait un traitement immunosuppresseur. Les patients atteints d'un CCR avec un contingent à cellules claires ont été randomisés (1:1) pour recevoir 200 mg de pembrolizumab toutes les 3 semaines (n = 496) ou un placebo (n = 498) pendant au maximum un an jusqu'à récidive de la maladie ou toxicité inacceptable. La randomisation a été stratifiée selon le statut métastatique (M0, M1 NED) et au sein du groupe M0, sous-stratifiée selon le statut de performance ECOG (0,1) et la région géographique (États-Unis, hors États-Unis). A partir de la randomisation, les patients ont bénéficié d'une imagerie toutes les 12 semaines pendant les 2 premières années, puis toutes les 16 semaines de la 3ème à la 5ème année, puis toutes les 24 semaines les années suivantes.
Parmi les 994 patients, les caractéristiques à l'inclusion étaient : âge médian de 60 ans (de 25 à 84 ans), 33 % âgés de 65 ans ou plus ; 71 % d'hommes ; et 85 % avaient un statut de performance ECOG de 0 et 15 % un statut de performance ECOG de 1. Quatre-vingt-quatorze pour cent étaient N0 ; 83 % n'avaient pas de composantes sarcomatoïdes ; 86 % étaient pT2 avec un Grade 4 ou des composantes sarcomatoïdes ou pT3 ; 8 % étaient pT4 ou avec une atteinte ganglionnaire ; et 6 % étaient M1 NED. Les caractéristiques à l'inclusion et les données démographiques étaient généralement comparables entre les bras pembrolizumab et placebo.
Le critère principal d'évaluation de l'efficacité était la survie sans maladie ou décès (DFS) évalué par l'investigateur. Le critère secondaire clé d'évaluation était l'OS. Lors de l'analyse intermédiaire pré-définie avec un suivi médian de 23,9 mois, l'étude a démontré une amélioration statistiquement significative de la DFS (HR 0,68 ; IC à 95 % 0,53-0,87 ; valeur de p = 0,0010) pour les patients randomisés dans le bras pembrolizumab par rapport au placebo. Les résultats d'efficacité actualisés avec un suivi médian de 29,7 mois sont résumés dans le Tableau 35 et la Figure 27.
Tableau 35 : Résultats d'efficacité dans KEYNOTE-564
| Critère d'évaluation | Pembrolizumab 200 mg toutes les 3 semainesn = 496 | Placebon = 498 |
| DFS | ||
| Nombre (%) de patients avec événement | 114 (23 %) | 169 (34 %) |
| Médiane en mois (IC 95%) | NA | NA |
| Hazard ratio* (IC 95%) | 0,63 (0,50 - 0,80) | |
| Valeur de p† | < 0,0001 | |
† Valeur de P nominale sur la base du test de Log rank stratifié NA = non atteint
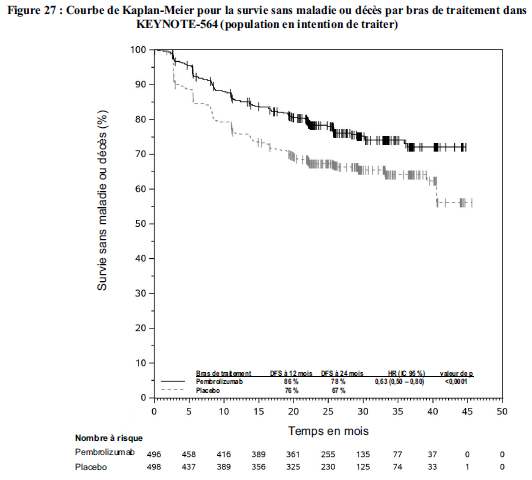
Au moment de l'analyse actualisée, le hazard ratio de la DFS (IC à 95 %) était de 0,68 (0,52 - 0,89) dans le sous-groupe de patients avec M0 à risque intermédiaire-élevé de récidive, 0,60 (0,33 - 1,10) dans le sous-groupe de patients avec M0 à risque élevé de récidive et 0,28 (0,12 - 0,66) dans le sous-groupe de patients avec M1 NED. Les résultats de l'OS n'étaient pas encore matures avec 23 décès sur 496 patients dans le bras pembrolizumab et 43 décès sur 498 patients dans le bras placebo.
Cancers MSI-H ou dMMR Cancer colorectal
KEYNOTE-177 : Etude contrôlée chez les patients atteints d'un cancer colorectal) MSI-H ou dMMR naïfs de traitement au stade métastatique
L'efficacité du pembrolizumab a été étudiée dans KEYNOTE-177, une étude multicentrique, randomisée, en ouvert, contrôlée par traitement actif, ayant inclus des patients atteints d'un cancer colorectal métastatique MSI-H ou dMMR non préalablement traité. Le statut tumoral MSI ou MMR (système de réparation des mésappariements de l'ADN/mismatch repair) a été déterminé localement par amplification en chaîne par polymérase (PCR) ou par IHC, respectivement. Les patients présentant une maladie auto-immune ou un état clinique nécessitant un traitement immunosuppresseur étaient inéligibles.
Les patients ont été randomisés (1:1) pour recevoir 200 mg de pembrolizumab par voie intraveineuse toutes les 3 semaines ou une des chimiothérapies suivantes au choix de l'investigateur, administrées par voie intraveineuse toutes les 2 semaines :
· mFOLFOX6 (oxaliplatine, leucovorine et FU) ou mFOLFOX6 en association avec bévacizumab ou cétuximab : oxaliplatine 85 mg/m2, leucovorine 400 mg/m2 (ou lévoleucovorine 200 mg/m2), et FU 400 mg/m2 en bolus le Jour 1, puis FU 2 400 mg/m2 sur 46-48 heures. Bévacizumab 5 mg/kg de poids corporel le Jour 1 ou cétuximab 400 mg/ m2 lors de la première perfusion puis 250 mg/m2 par semaine.· FOLFIRI (irinotecan, leucovorine et FU) ou FOLFIRI en association avec bévacizumab ou cétuximab : irinotécan 180 mg/m2, leucovorine 400 mg/m2 (ou lévoleucovorine 200 mg/m2), et FU 400 mg/m2 en bolus le Jour 1, puis FU 2 400 mg/m2 sur 46-48 heures. Bévacizumab 5 mg/kg de poids corporel le Jour 1 ou cétuximab 400 mg/m2 lors de la première perfusion puis 250 mg/m2 par semaine.
Le traitement par pembrolizumab a été poursuivi jusqu'à progression de la maladie définie par RECIST v1.1 telle que déterminée par l'investigateur, ou toxicité inacceptable. Les patients traités par pembrolizumab sans progression de la maladie pouvaient être traités jusqu'à 24 mois. Une évaluation du statut tumoral était réalisée toutes les 9 semaines. Les patients randomisés pour recevoir la chimiothérapie pouvaient bénéficier de pembrolizumab au moment de la progression de la maladie.
Un total de 307 patients ont été inclus et randomisés pour recevoir pembrolizumab (n = 153) ou une chimiothérapie (n = 154). Les caractéristiques de ces patients à l'inclusion étaient : âge médian de 63 ans (24 à 93 ans) ; 47 % âgés de 65 ans ou plus ; 50 % d'hommes ; 75 % de type caucasien et 16 % de type asiatique ; 52 % et 48 % avaient un statut de performance ECOG de 0 ou 1, respectivement. Statut mutationnel : 25 % BRAF V600E, 24 % KRAS/NRAS. Parmi les 143 patients traités par chimiothérapie, 56 % recevaient mFOLFOX6 avec ou sans bévacizumab ou cétuximab et 44 % recevaient FOLFIRI avec ou sans bévacizumab ou cétuximab.
Les critères principaux d'évaluation de l'ef icacité étaient la PFS (telle qu'évaluée en aveugle par une revue centralisée indépendante utilisant RECIST v1.1) et l'OS. Les critères secondaires d'évaluation étaient l'ORR et la durée de réponse. L'étude a démontré une amélioration statistiquement significative de la PFS (HR 0,60 ; IC à 95 % 0,45-0,80 ; valeur de p 0,0002) pour les patients randomisés dans le bras pembrolizumab par rapport à la chimiothérapie lors de l'analyse finale prédéfinie de la PFS. Il n'y avait pas de différence statistiquement significative entre le pembrolizumab et la chimiothérapie dans l'analyse finale de l'OS dans laquelle 60 % des patients qui avaient été randomisés pour recevoir une chimiothérapie avaient reçu en cross-over des traitements anti-PD-1/PD-L1 ultérieurs, y compris le pembrolizumab. Le tableau 36 résume les critères clés d'efficacité et les figures 28 et 29 montrent les courbes de Kaplan Meier pour la PFS et l'OS mises à jour sur la base de l'analyse finale avec un suivi médian de 38,1 mois (de 0,2 à 58,7 mois).
Tableau 36 : Résultats d'efficacité dans KEYNOTE-177
| Critère d'évaluation | Pembrolizumab 200 mg toutes les 3 semainesn = 153 | Chimiothérapie n = 154 |
| PFS* | ||
| Nombre (%) de patients avec événement | 86 (56 %) | 117 (76 %) |
| Médiane en mois (IC 95 %) | 16,5 (5,4 - 38,1) | 8,2 (6,1- 10,2) |
| Hazard ratio† (IC 95 %) | 0,59 (0,45 - 0,79) | |
| Value de p‡ | 0,0001 | |
| OS§ | ||
| Nombre (%) de patients avec événement | 62 (41 %) | 78 (51 %) |
| Médiane en mois (IC 95 %) | NA (49,2 - NA) | 36,7 (27,6 - NA) |
| Hazard ratio† (IC 95 %) | 0,74 (0,53 - 1,03) | |
| Valeur de p§ | 0,0359 | |
| Taux de réponse objective | ||
| ORR % (IC 95 %) | 45 % (37,1 - 53,3) | 33 % (25,8 - 41,1) |
| Réponse complète | 13 % | 4 % |
| Réponse partielle | 32 % | 29 % |
| Durée de réponse | ||
| Médiane en mois (intervalle) | NA (2,3+ - 53,5+) | 10,6 (2,8 - 48,3+) |
| % avec durée ≥ 24 mois¶ | 84 % | 34 % |
† Sur la base du modèle “Cox regression »
‡ La valeur de p est nominale.
§ Non statistiquement significatif après ajustement pour multiplicité
¶ Sur la base de l'estimation de Kaplan-Meier NA = Non atteint
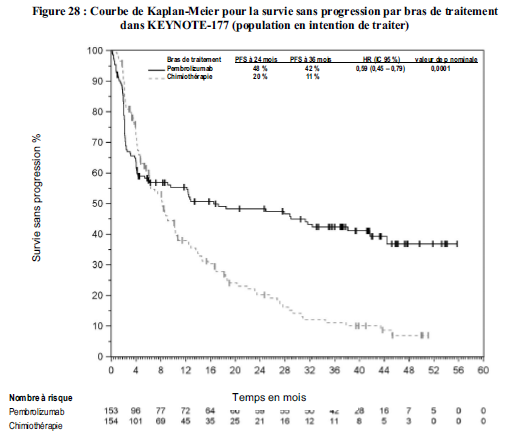
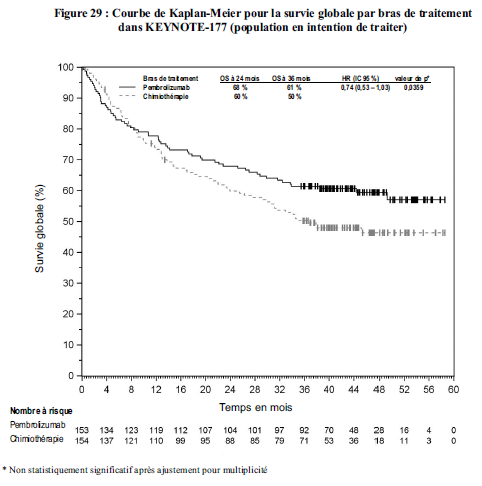
KEYNOTE-164 : Etude en ouvert chez les patients atteints d'un cancer colorectal MSI-H ou dMMR non résécable ou métastatique ayant reçu un traitement antérieur
L'efficacité du pembrolizumab a été étudiée dans KEYNOTE-164, une étude de phase II, multicentrique, non randomisée, en ouvert, multi cohortes, ayant inclus des patients atteints d'un cancer colorectal MSI-H ou dMMR non résécable ou métastatique ayant progressé après un traitement antérieur à base de fluoropyrimidine en association à l'irinotécan et/ou l'oxaliplatine. Les patients ont reçu 200 mg de pembrolizumab toutes les 3 semaines jusqu'à toxicité inacceptable ou progression de la maladie. Les patients cliniquement stables avec une preuve initiale de progression de la maladie pouvaient rester sous traitement jusqu'à ce que la progression de la maladie soit confirmée. Les patients sans progression de leur maladie étaient traités jusqu'à 24 mois (jusqu'à 35 cycles). Une évaluation du statut tumoral était réalisée toutes les 9 semaines.
Parmi les 124 patients inclus dans KEYNOTE-164, les caractéristiques à l'inclusion étaient : âge médian de 56 ans (35 % âgés de 65 ans ou plus) ; 56 % d'hommes ; 68 % de type Caucasien, 27 % de type Asiatique, 41 % et 59 % avaient un statut de performance ECOG de 0 et 1, respectivement. Douze pour cent des patients présentaient des mutations BRAF et 36 % présentaient des mutations RAS ; 39 % et 34 % étaient indéterminés pour les mutations BRAF et RAS, respectivement. Quatre-vingt-dix-sept pour cent des patients avaient une maladie au stade M1 et 3 % avaient une maladie au stade M0 (localement avancé non résécable). Soixante-seize pour cent des patients ont reçu au moins 2 lignes de traitement antérieures.
Le critère principal d'évaluation de l'efficacité était l'ORR tel qu'évalué en aveugle par une revue centralisée indépendante (BICR) utilisant RECIST 1.1. Les critères secondaires d'évaluation de l'efficacité incluaient la durée de réponse, la PFS et l'OS. La durée médiane de suivi en mois était de 37,3 (intervalle : 0,1 à 65,2).
Les résultats d'efficacité sont résumés dans le tableau 37.
Tableau 37 : résultats d'efficacité dans l'étude KEYNOTE-164
| Critère d'évaluation | n=124 |
| Taux de réponse objective* | |
| ORR % (IC 95 %) | 34 % (25,6 ; 42,9) |
| Réponse complète | 10 % |
| Réponse partielle | 24 % |
| Durée de réponse* | |
| Médiane en mois (intervalle) | NA (4,4 ; 58,5+) |
| % avec une durée ≥ 36 mois# | 92 % |
# Sur la base de l'estimation Kaplan-Meier
+ Indique qu'il n'y pas de maladie évolutive au moment de la dernière évaluation de la maladie NA = non atteint
Des réponses objectives ont été observées quel que soit le statut de mutation BRAF ou RAS.
Cancers non-colorectaux
KEYNOTE-158 : Etude en ouvert chez les patients atteints d'un cancer MSI-Hou dMMR de l'endomètre, de l'estomac, de l'intestin grêle ou des voies biliaires non résécable ou métastatique ayant reçu un traitement antérieur
L'efficacité du pembrolizumab a été étudiée chez 355 patients atteints de tumeurs solides MSI-H ou dMMR non résécables ou métastatiques hors cancer colorectal, inclus dans une étude de phase II (KEYNOTE-158), multicentrique, non randomisée, en ouvert, incluant des patients atteints d'un cancer de l'endomètre, de l'estomac, de l'intestin grêle ou des voies biliaires. Le statut tumoral MSI ou MMR a été déterminé prospectivement par PCR ou IHC, respectivement.
Les patients ont reçu 200 mg de pembrolizumab toutes les 3 semaines jusqu'à toxicité inacceptable ou progression de la maladie. Les patients cliniquement stables avec une preuve initiale de progression de la maladie pouvaient rester sous traitement jusqu'à ce que la progression de la maladie soit confirmée. Les patients sans progression de leur maladie étaient traités jusqu'à 24 mois (jusqu'à 35 cycles). Une évaluation du statut tumoral était réalisée toutes les 9 semaines pendant la première année, puis toutes les 12 semaines par la suite.
Parmi les 83 patientes atteintes d'un cancer de l'endomètre, les caractéristiques à l'inclusion étaient : âge médian de 64 ans (intervalle : 42 à 86) ; 46 % âgées de 65 ans ou plus ; 84 % de type caucasien, 6 % de type asiatique et 4 % de type noir ; et un statut de performance ECOG de 0 (46 %) et 1 (54 %). Quatre-vingt-dix- huit pour cent des patientes avaient une maladie au stade M1 et 2 % avaient une maladie au stade M0.
Quarante-sept pour cent des patients ont reçu au moins 2 lignes de traitement antérieures.
Parmi les 51 patients atteints d'un cancer gastrique, les caractéristiques à l'inclusion étaient : âge médian de 67 ans (intervalle : 41 à 89) ; 57 % âgés de 65 ans ou plus ; 65 % d'hommes, 63 % de type caucasien, 28 % de type asiatique ; et un statut de performance ECOG de 0 (45 %) et 1 (55 %). Tous les patients avaient une maladie au stade M1. Quarante-cinq pour cent des patients ont reçu au moins 2 lignes de traitement antérieures.
Parmi les 27 patients atteints d'un cancer de l'intestin grêle, les caractéristiques à l'inclusion étaient : âge médian de 58 ans (intervalle : 21 à 77) ; 33 % âgés de 65 ans ou plus ; 63 % d'hommes, 81 % de type caucasien, 11 % de type asiatique ; et un statut de performance ECOG de 0 (56 %) et 1 (44 %). Quatre- vingt-seize pour cent des patients avaient une maladie au stade M1 et 4 % avaient une maladie au stade M0. Trente-sept pour cent des patients ont reçu au moins 2 lignes de traitement antérieures. Tous les patients avaient une histologie tumorale d'adénocarcinome.
Parmi les 22 patients atteints d'un cancer des voix biliaires, les caractéristiques à l'inclusion étaient : âge médian de 61 ans (intervalle : 40 à 77) ; 41 % âgés de 65 ans ou plus ; 73 % d'hommes, 91 % de type caucasien, 9 % de type asiatique ; un statut de performance ECOG de 0 (45 %) et 1 (55 %) ; et 82 % avaient une maladie au stade M1 et 18 % avaient une maladie au stade M0. Quarante et un pour cent des patients ont reçu au moins 2 lignes de traitement antérieures.
Le critère principal d'évaluation de l'efficacité était l'ORR tel qu'évalué en aveugle par une revue centralisée indépendante (BICR) utilisant RECIST 1.1. Les critères secondaires d'évaluation de l'ef icacité incluaient la durée de réponse, la PFS et l'OS. La durée médiane de suivi en mois était de 21,9 (intervalle : 1,5 à 64,0) pour le cancer de l'endomètre, 13,9 (intervalle : 1,1 à 66,9) pour le cancer gastrique, 29,1 (4,2 à 67,7) pour le cancer de l'intestin grêle et 19,4 (intervalle : 1,1 à 60,8) pour le cancer des voies biliaires. Les résultats d'efficacité sont résumés dans le tableau 38.
Tableau 38 : résultats d'efficacité dans l'étude KEYNOTE-158
| Critère d'évaluation | Endomètre n = 83 | Gastrique n = 51 | Intestin grêle n = 27 | Voies biliairesn = 22 |
| Taux de réponse objective* | ||||
| ORR % | 51 % | 37 % | 56 % | 41 % |
| (IC 95 %) | (39,4 ; 61,8) | (24,1 ; 51,9) | (35,3 ; 74,5) | (20,7 ; 63,6) |
| Réponse complète | 16 % | 14 % | 15 % | 14 % |
| Réponse partielle | 35 % | 24 % | 41 % | 27 % |
| Durée de réponse* | ||||
| Médiane en mois (intervalle) | NA | NA | NA | 30,6 |
| (2,9 ; 60,4+) | (6,2 ; 63,0+) | (3,7+ ; 57,3+) | (6,2 ; 46,0+) | |
| % avec durée ≥ 12 mois# | 85 % | 90 % | 93 % | 89 % |
| % avec durée ≥ 36 mois# | 60 % | 81 % | 73 % | 42 % |
# Sur la base de l'estimation Kaplan-Meier
+ Indique qu'il n'y pas de maladie évolutive au moment de la dernière évaluation de la maladie
NA = non atteint
Cancer de l'œsophage
KEYNOTE-590 : Étude contrôlée du traitement en association chez les patients atteints d'un cancer de l'œsophage naïfs de traitement
L'efficacité du pembrolizumab en association à une chimiothérapie a été étudiée dans KEYNOTE-590, une étude multicentrique, randomisée, en double aveugle, contrôlée versus placebo chez des patients atteints d'un cancer de l'œsophage ou d'un adénocarcinome de la jonction gastro-œsophagienne (Siewert type I) localement avancés non résécables ou métastatiques. Les patients atteints d'une maladie auto-immune active, dont l'état clinique nécessitait un traitement immunosuppresseur, ou les patients atteints d'un adénocarcinome de la jonction gastro-œsophagienne HER-2 positif connu, étaient inéligibles pour l'étude. La randomisation était stratifiée selon l'histologie de la tumeur (carcinome épidermoïde vs adénocarcinome), la région géographique (Asie vs hors Asie) et le statut de performance ECOG (0 vs 1).
Les patients ont été randomisés (1 : 1) dans l'un des bras de traitement suivants :
· Pembrolizumab 200 mg le Jour 1 de chaque cycle de trois semaines en association au cisplatine 80 mg/m2 IV le Jour 1 de chaque cycle de trois semaines jusqu'à six cycles et 5-FU 800 mg/m2 IV par jour du Jour 1 au Jour 5 de chaque cycle de trois semaines, ou selon la norme locale pour l'administration de 5-FU.· Placebo le Jour 1 de chaque cycle de trois semaines en association au cisplatine 80 mg/m2 IV le Jour 1 de chaque cycle de trois semaines jusqu'à six cycles et 5-FU 800 mg/m2 IV par jour du Jour 1 au Jour 5 de chaque cycle de trois semaines, ou selon la norme locale pour l'administration de 5-FU.
Le traitement par pembrolizumab ou chimiothérapie était poursuivi jusqu'à progression de la maladie ou toxicité inacceptable ou une durée maximale de 24 mois. Si cliniquement stables, les patients randomisés pour recevoir le pembrolizumab étaient autorisés à continuer au-delà de la première progression de la maladie définie selon RECIST v1.1, jusqu'à ce que les premières preuves radiographiques de la progression de la maladie soient confirmées par une nouvelle imagerie réalisée au moins 4 semaines plus tard. Une évaluation du statut tumoral était réalisée toutes les 9 semaines.
Parmi les 749 patients de l'étude KEYNOTE-590, 383 (51 %) avaient des tumeurs qui exprimaient PD-L1 avec un CPS ≥ 10 avec le kit PD-L1 IHC 22C3 pharmDxTM.. Les caractéristiques à l'inclusion de ces 383 patients étaient : âge médian de 63 ans (de 28 à 89 ans), 41 % âgés de 65 ans ou plus ; 82 % d'hommes ; 34 % de type Caucasien et 56 % de type Asiatique ; 43 % et 57 % avaient respectivement un statut de performance ECOG de 0 ou 1. Quatre-vingt-treize pour cent présentaient une maladie au stade M1. Soixante- quinze pour cent avaient une histologie tumorale de type carcinome épidermoïde et 25 % de type adénocarcinome.
Les critères principaux d'évaluation de l'ef icacité étaient l'OS et la PFS tels qu'évalués par l'investigateur selon RECIST 1.1 dans les populations avec une histologie épidermoïde, CPS ≥ 10, et chez tous les patients. L'étude a démontré une amélioration statistiquement significative de l'OS et de la PFS pour toutes les populations pré-définies de l'étude. Chez tous les patients randomisés dans le groupe pembrolizumab en association à une chimiothérapie, par rapport à la chimiothérapie, le HR de l'OS était de 0,73 (IC 95 %, 0,62 - 0,86) et le HR de la PFS était de 0,65 (IC 95 %, 0,55 - 0,76). Les critères secondaires d'évaluation de l'efficacité étaient l'ORR et la durée de réponse, tel qu'évalués par l'investigateur selon RECIST 1.1. Le tableau 39 résume les critères clés d'efficacité de l'analyse pré-définie dans KEYNOTE-590, réalisée après un suivi médian de 13,5 mois (de 0,5 à 32,7 mois) chez les patients dont les tumeurs exprimaient PD-L1 avec un CPS ≥ 10. Les courbes de Kaplan-Meier pour l'OS et la PFS sont présentées dans les figures 30 et 31.
Tableau 39 : Résultats d'ef icacité pour pembrolizumab plus chimiothérapie dans KEYNOTE-590 avec expression de PD-L1 (CPS ≥ 10)
| Critère d'évaluation | Pembrolizumab Chimiothérapie à base de cisplatine5-FUn = 186 | Traitement standard* n = 197 |
| OS | ||
| Nombre (%) de patients avec événement | 124 (66,7 %) | 165 (83,8 %) |
| Médiane en mois† (IC 95 %) | 13,5 (11,1 - 15,6) | 9,4 (8,0 - 10,7) |
| Hazard ratio‡ (IC 95 %) | 0,62 (0,49 - 0,78) | |
| Valeur de p§ | < 0,0001 | |
| PFS¶ | ||
| Nombre (%) de patients avec événement | 140 (75,3 %) | 174 (88,3 %) |
| Médiane en mois† (IC 95 %) | 7,5 (6,2 - 8,2) | 5,5 (4,3 - 6,0) |
| Hazard ratio‡ (IC 95 %) | 0,51 (0,41 - 0,65) | |
| Valeur de p§ | < 0,0001 | |
| Taux de réponse objective¶ | ||
| ORR§ % (IC 95 %) | 51,1 (43,7 - 58,5) | 26,9 (20,8 - 33,7) |
| Réponse complète | 5,9 % | 2,5 % |
| Réponse partielle | 45,2 % | 24,4 % |
| Valeur de p# | < 0,0001 | |
| Durée de réponse¶,Þ | ||
| Médiane en mois (intervalle) | 10,4 (1,9 - 28,9+) | 5,6 (1,5+ - 25,0+) |
| % avec durée ≥ 6 mois† | 80,2 % | 47,7 % |
| % avec durée ≥ 12 mois† | 43,7 % | 23,2 % |
| % avec durée ≥ 18 mois† | 33,4 % | 10,4 % |
† Sur la base de l'estimation de Kaplan Meier
‡ Sur la base du modèle « Cox proportional hazard » stratifié
§ Valeur de p unilatérale sur la base du test de log-rank stratifié par région géographique (Asie versus le Reste du Monde) et histologie de la tumeur (Adénocarcinome versus Carcinome Epidermoïde) et indice de performance ECOG (0 versus 1)
¶ Évalué par l'investigateur selon RECIST 1.1
# Valeur de p unilatérale pour les tests. H0 : différence en % = 0 versus H1 : différence en % > 0
Þ Meilleure réponse objective telle que réponse complète confirmée ou réponse partielle.
Un total de 32 patients âgés de ≥ 75 ans avec un CPS PD-L1 ≥ 10 ont été inclus dans l'étude KEYNOTE-590 (18 dans le groupe pembrolizumab en association et 14 dans le groupe contrôle). Les données sur l'efficacité du pembrolizumab en association à la chimiothérapie sont trop limitées dans cette population de patients.
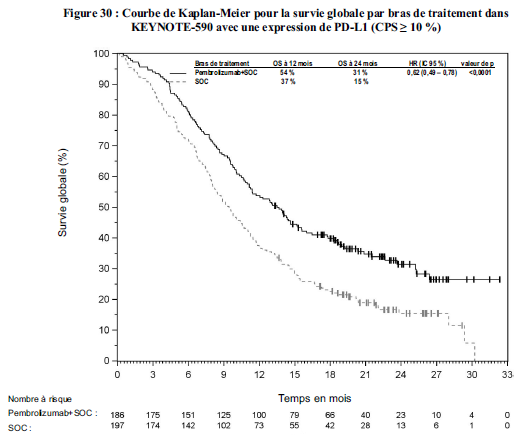
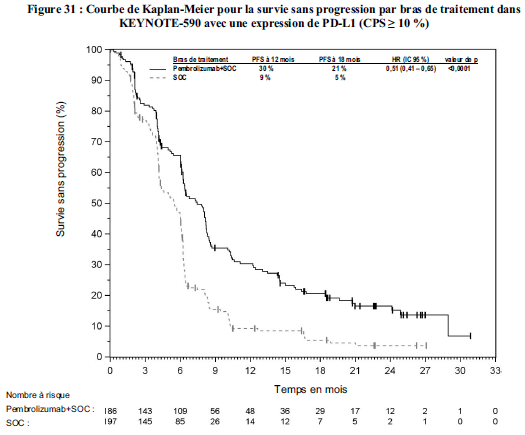
Cancer du sein triple négatif
KEYNOTE-522 : Étude contrôlée du traitement néoadjuvant et adjuvant chez les patients atteints d'un cancer du sein triple négatif localement avancé, inflammatoire ou de stade précoce à haut risque de récidive
L'efficacité du pembrolizumab en association à une chimiothérapie comme traitement néoadjuvant, puis poursuivi après la chirurgie en monothérapie comme traitement adjuvant a été étudiée dans KEYNOTE-522, une étude randomisée, en double aveugle, multicentrique, contrôlée versus placebo. Si indiqué, les patients avaient reçu une radiothérapie adjuvante avant ou pendant le traitement adjuvant par pembrolizumab ouplacebo. Les principaux critères d'éligibilité pour cette étude étaient les CSTN localement avancés, inflammatoires ou de stade précoce à haut risque de récidive (taille tumorale > 1 cm mais ≤ 2 cm de diamètre avec atteinte ganglionnaire ou taille tumorale > 2 cm de diamètre indépendamment de l'atteinte ganglionnaire), quelle que soit l'expression tumorale de PD-L1. Les patients atteints d'une maladie auto-immune active ayant nécessité un traitement systémique dans les 2 ans précédents le traitement ou dont l'état clinique nécessitait un traitement immunosuppresseur étaient inéligibles pour l'étude. La randomisation était stratifiée selon le statut ganglionnaire (positif vs négatif), la taille de la tumeur (T1/T2 vs T3/T4) et le choix du carboplatine (administré toutes les 3 semaines vs chaque semaine). Les patients étaient randomisés (2:1) pour recevoir soit du pembrolizumab, soit du placebo en perfusion intraveineuse :
· Quatre cycles de pembrolizumab 200 mg ou de placebo toutes les 3 semaines en néoadjuvant le Jour 1 des cycles 1-4 du schéma thérapeutique en association au :
§ Carboplatine
§ ASC
5 mg/mL/min toutes les 3 semaines le Jour 1 des cycles 1-4 du schéma
thérapeutique
ou ASC 1,5 mg/mL/min chaque semaine les Jours 1, 8 et 15 des cycles 1-4 du schéma thérapeutique et
§ Paclitaxel 80 mg/m2 chaque semaine les Jours 1, 8 et 15 des cycles 1-4 du schéma thérapeutique
· Suivi de quatre cycles supplémentaires de pembrolizumab 200 mg ou de placebo toutes les 3 semaines en néoadjuvant le Jour 1 des cycles 5-8 du schéma thérapeutique en association au :
§ Doxorubicine 60 mg/m2ou épirubicine 90 mg/m2 toutes les 3 semaines le Jour 1 des cycles 5-8 du schéma thérapeutique et
· Cyclophosphamide
600 mg/m2 toutes les 3 semaines le Jour 1 des cycles 5-8 du schéma thérapeutique
· Après
la chirurgie, 9 cycles de pembrolizumab 200 mg ou de placebo toutes les 3
semaines en adjuvant ont été administrés.
Le traitement par pembrolizumab ou placebo était poursuivi jusqu'à la fin du traitement (17 cycles), progression de la maladie empêchant une chirurgie définitive, récidive de la maladie en phase adjuvante ou toxicité inacceptable.
Un total de 1 174 patients a été randomisé. Les caractéristiques de la population de l'étude étaient : âge médian de 49 ans (de 22 à 80) ; 11 % âgés de 65 ans ou plus ; 99,9 % de femmes ; 64 % de type Caucasien ; 20 % de type Asiatique, 5 % de type Noir et 2 % de type Indien d'Amérique ou d'Alaska ; le statut de performance ECOG de 0 (87 %) et 1 (13 %) ; 56 % étaient en état de pré-ménopause et 44 % étaient en état de post-ménopause ; 7 % étaient des Tumeurs primitives 1 (T1), 68 % T2, 19 % T3 et 7 % T4 ; 49 % étaient une atteinte ganglionnaire 0 (N0), 40 % N1, 11 % N2 et 0,2 % N3 ; 1,4 % des patients avaient un cancer du sein inflammatoire ; 75 % des patients étaient principalement au stade II et 25 % au stade III.
Les deux critères principaux d'évaluation de l'efficacité étaient le taux de réponse histologique complète (pCR) et la survie sans événement (EFS). La pCR a été définie comme l'absence de cancer invasif dans le sein et les ganglions lymphatiques (ypT0/Tis ypN0) et a été évaluée en aveugle par le pathologiste local au moment de la chirurgie définitive. L'EFS a été définie comme le délai entre la randomisation et la première apparition de l'un des événements suivants : progression de la maladie qui empêche une chirurgie définitive, récidive locale ou à distance, deuxième tumeur maligne primitive ou décès quelle qu'en soit la cause. L'étude a démontré une amélioration statistiquement significative de la différence de taux de pCR lors de son analyse primaire prédéfinie (n = 602), les taux de pCR étaient de 64,8 % (IC 95 % : 59,9 % - 69,5 %) dans le bras pembrolizumab et 51,2 % (IC 95 % : 44,1 % - 58,3 %) dans le bras placebo, avec une différence de traitement de 13,6 % (IC 95 % : 5,4 % - 21,8 % ; valeur de p 0,00055). L'étude a également démontré une amélioration statistiquement significative de l'EFS lors de son analyse prédéfinie. Le critère secondaire d'évaluation de l'efficacité était l'OS. Au moment de l'analyse de l'EFS, les résultats de l'OS n'étaient pas encore matures (45 % des événements requis pour l'analyse finale). Lors d'une analyse intermédiaire prédéfinie, la durée de suivi médiane pour tous les patients était de 37,8 mois (de 2,7 à 48 mois). Le tableau 40 résume les critères clés d'efficacité des analyses prédéfinies. Les courbes de Kaplan-Meier pour l'EFS et l'OS sont représentées en figures 32 et 33.
Tableau 40 : Résultats d'efficacité dans KEYNOTE-522
| Critère d'évaluation | Pembrolizumab avec Chimiothérapie/Pembrolizumab | Placebo avec Chimiothérapie/Placebo |
| pCR (ypT0/Tis ypN0)* | n = 669 | n = 333 |
| Nombre de patients avec pCR | 428 | 182 |
| Taux (%) de pCR (IC 95 %) | 64,0 (60,2 ; 67,6) | 54,7 (49,1 ; 60,1) |
| Estimation (%) de la différence de traitement (IC 95 %)† | 9,2 (2,8 ; 15,6) | |
| Valeur p‡ | 0,00221 | |
| EFS§ | n = 784 | n = 390 |
| Nombre (%) de patients avec événement | 123 (15,7 %) | 93 (23,8 %) |
| EFS à 24 mois (IC 95 %) | 87,8 (85,3 ; 89,9) | 81,0 (76,8 ; 84,6) |
| Hazard ratio (IC 95 %)¶ | 0,63 (0,48 ; 0,82) | |
| Valeur p# | 0,00031 | |
| OSÞ | ||
| Nombre (%) de patients avec événement | 80 (10,2 %) | 55 (14,1 %) |
| OS à 24 mois (IC 95 %) | 92,3 (90,2 ; 94,0) | 91,0 (87,7 ; 93,5) |
| Hazard ratio (IC 95 %)¶ | 0,72 (0,51 ; 1,02) | |
† Basé sur la méthode de Miettinen et Nurminen stratifiée par statut ganglionnaire, taille de la tumeur, et choix du carboplatine
‡ Valeur de p unilatérale pour les tests. H0 : différence en % = 0 versus H1 : différence en % > 0
§ Sur la base d'une analyse intermédiaire prédéfinie de l'EFS (par rapport à un niveau de significativité de 0,0052)
¶ Basé sur le modèle de régression de Cox avec la méthode d'Efron pour gérer les ex-aequo avec traitement comme covariable stratifiée par statut ganglionnaire, taille de la tumeur, et choix du carboplatine
# Valeur de p unilatérale sur la base du test du log rank stratifié par statut ganglionnaire, taille de la tumeur, et choix du carboplatine
Þ Les résultats de l'OS lors de l'analyse intermédiaire n'ont pas atteint la limite d'ef icacité prédéfinie de 0,00085861 pour la significativité statistique.
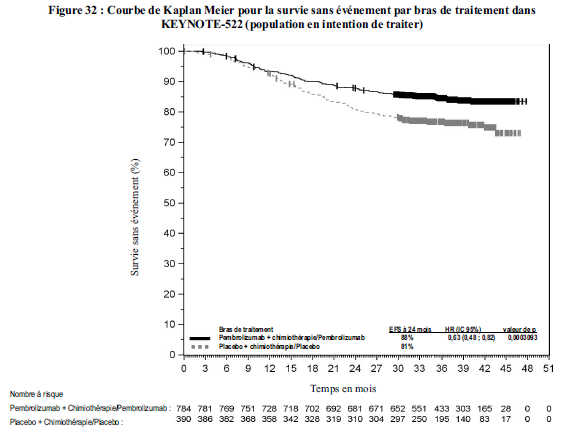
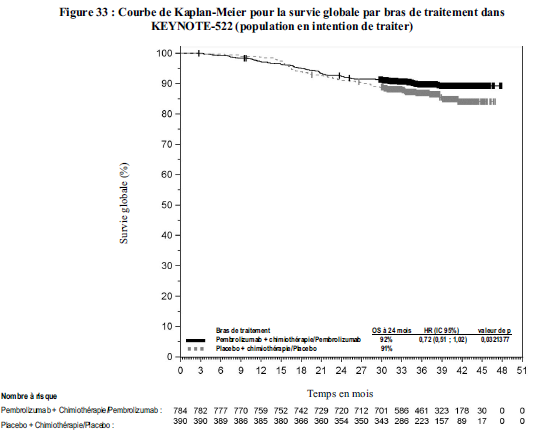
KEYNOTE-355 : Étude contrôlée du traitement en association chez les patients atteints d'un CSTN non préalablement traités pour la maladie métastatique
L'efficacité du pembrolizumab en association au paclitaxel, nab-paclitaxel ou gemcitabine et carboplatine a été étudiée dans KEYNOTE-355, une étude multicentrique, randomisée, en double aveugle, contrôlée versus placebo. Les principaux critères d'éligibilité étaient un CSTN localement récurrent non résécable ou métastatique, quel que soit le statut d'expression tumorale de PD-L1, non préalablement traité par chimiothérapie au stade avancé. Les patients présentant une maladie auto-immune active ayant nécessité un traitement systémique dans les 2 ans précédents le traitement ou un état clinique nécessitant un traitement immunosuppresseur étaient inéligibles. La randomisation était stratifiée selon la chimiothérapie (paclitaxel ou nab-paclitaxel vs gemcitabine et carboplatine), le statut d'expression tumorale de PD-L1 (CPS ≥ 1 vs CPS < 1) et le traitement antérieur par une chimiothérapie de la même classe au stade néoadjuvant (oui vs. non). Les patients ont été randomisés (2 : 1) dans l'un des bras de traitement par perfusion intraveineuse suivants:
· Pembrolizumab 200 mg le Jour 1 toutes les 3 semaines en association au nab-paclitaxel 100 mg/m2 le Jour 1, 8 et 15 tous les 28 jours, ou au paclitaxel 90 mg/m2 le Jour 1, 8 et 15 tous les 28 jours, ou à la gemcitabine 1 000 mg/m2 et au carboplatine ASC 2 mg/mL/min le Jour 1 et 8 tous les 21 jours.· Placebo le Jour 1 toutes les 3 semaines en association au nab-paclitaxel 100 mg/m2 le Jour 1, 8 et 15 tous les 28 jours, ou au paclitaxel 90 mg/m2 le Jour 1, 8 et 15 tous les 28 jours, ou à la gemcitabine 1 000 mg/m2 et au carboplatine ASC 2 mg/mL/min le Jour 1 et 8 tous les 21 jours.
Le traitement par pembrolizumab ou placebo, tous deux en association à une chimiothérapie, était poursuivi jusqu'à progression de la maladie définie par RECIST 1.1 telle que déterminée par l'investigateur, toxicité inacceptable ou une durée maximale de 24 mois. La chimiothérapie pouvait se poursuivre selon le traitement standard. L'administration de pembrolizumab était autorisée au-delà de la progression de la maladie définie par RECIST si le patient était cliniquement stable et si l'investigateur considérait qu'il y avait un bénéfice clinique. Une évaluation de la réponse tumorale était réalisée à la Semaine 8, 16 et 24, et ensuite toutes les 9 semaines pendant la première année, puis toutes les 12 semaines par la suite.
Parmi les 847 patients randomisées de l'étude KEYNOTE-355, 636 (75 %) avaient des tumeurs qui exprimaient PD-L1 avec un CPS ≥ 1 et 323 (38 %) avaient une expression tumorale de PD-L1 avec un CPS ≥ 10 avec le kit PD-L1 IHC 22C3 pharmDxTM.. Les caractéristiques à l'inclusion de ces 323 patients dont les tumeurs exprimaient PD-L1 avec un CPS ≥ 10 comprenaient : âge médian de 53 ans (de 22 à 83 ans), 20 % âgés de 65 ans ou plus ; 100 % de femmes ; 69 % de type Caucasien, 20 % de type Asiatique et 5 % de type Noir ; un statut de performance ECOG de 0 (61 %) et 1(39 %) ; 67 % étaient en post- ménopause ; 3 % avaient des antécédents de métastases cérébrales ; et 20 % présentaient un intervalle sans maladie < 12 mois.
Les critères principaux d'évaluation de l'ef icacité étaient la PFS telle qu'évaluée en aveugle par une revue centralisée indépendante utilisant RECIST 1.1 et l'OS. Les critères secondaires d'évaluation de l'ef icacité étaient l'ORR et la durée de réponse, tels qu'évalués en aveugle par une revue centralisée indépendante utilisant RECIST 1.1. L'étude a démontré une amélioration statistiquement significative de la PFS lors de son analyse intermédiaire prédéfinie (HR 0,65 ; IC 95 % 0,49-0,86; valeur de p 0,0012) et de l'OS lors de l'analyse finale chez les patients dont les tumeurs exprimaient PD-L1 avec un CPS ≥ 10, randomisés dans le bras pembrolizumab en association avec la chimiothérapie par rapport au placebo en association avec la chimiothérapie.
Le tableau 41 résume les critères clés d'efficacité et les figures 34 et 35 représentent les courbes de Kaplan- Meier pour la PFS et l'OS, basées sur l'analyse finale avec un temps de suivi médian de 20,2 mois (intervalle : 0,3 à 53,1 mois) pour les patients dont les tumeurs exprimaient PD-L1 avec un CPS ≥ 10.
Tableau 41 : Résultats d'ef icacité chez les patients avec CPS ≥ 10 dans KEYNOTE-355
| Critère d'évaluation | Pembrolizumab avec chimiothérapie*n = 220 | Placebo avec chimiothérapie*n = 103 |
| PFS† | ||
| Nombre (%) de patients avec événement | 144 (65 %) | 81 (79 %) |
| Hazard ratio‡ (IC 95 %) | 0,66 (0,50 - 0,88) | |
| Valeur de p§ | 0,0018 | |
| Médiane en mois (IC 95 %) | 9,7 (7,6 - 11,3) | 5,6 (5,3 - 7,5) |
| OS | ||
| Nombre (%) de patients avecévénement | 155 (70 %) | 84 (82 %) |
| Hazard ratio‡ (IC 95 %) | 0,73 (0,55 - 0,95) | |
| Valeur de p¶ | 0,0093 | |
| Médiane en mois (IC 95 %) | 23,0 (19,0 - 26,3) | 16,1 (12,6 - 18,8) |
| Taux de réponse objective† | ||
| ORR % (IC 95 %) | 53 % (46 - 59) | 41 % (31 - 51) |
| Réponse complète | 17 % | 14 % |
| Réponse partielle | 35 % | 27 % |
| Durée de réponse† | ||
| Médiane en mois (intervalle) | 12,8 (1,6+ - 45,9+) | 7,3 (1,5 - 46,6+) |
| % de patients avec durée ≥ 6 mois# | 82 % | 60 % |
| % de patients avec durée ≥ 12 mois# | 56 % | 38 % |
† Évalué en aveugle par une revue centralisée indépendante utilisant RECIST 1.1
‡ Basé sur le modèle de régression de Cox avec la méthode d'Efron pour gérer les ex-aequo avec traitement comme covariable stratifiée par chimiothérapie dans l'étude (taxane vs. gemcitabine et carboplatine) et traitement antérieur par une chimiothérapie de la même classe au stade néoadjuvant (oui vs. non)
§ Valeur de p nominale sur la base du test de log-rank stratifié par chimiothérapie dans l'étude (taxane vs gemcitabine et carboplatine) et traitement antérieur par une chimiothérapie de la même classe au stade néoadjuvant (oui vs. non). Lors de l'analyse intermédiaire prédéfinie de la PFS (durée de suivi médiane de 19,2 mois), une supériorité statistiquement significative a été atteinte pour la PFS en comparant pembrolizumab/chimiothérapie avec placebo/chimiothérapie Valeur de p de 0,0012.
¶ Valeur de p unilatérale basée sur un test du log-rank stratifié par chimiothérapie dans l'étude (taxane vs gemcitabine et carboplatine) et traitement antérieur par une chimiothérapie de la même classe au stade néoadjuvant (oui vs non). Les résultats de l'OS ont atteint la limite d'ef icacité prédéfinie de 0,0113 pour la significativité statistique.
# A partir de la méthode du produit limite (Kaplan-Meier) pour les données censurées
+ Indique qu'il n'y a pas de maladie évolutive au moment de la dernière évaluation de la maladie
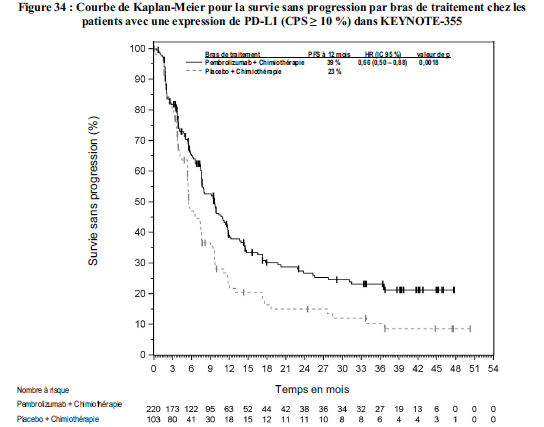
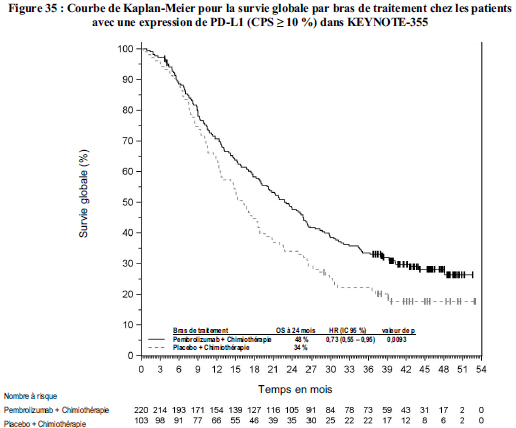
Cancer de l'endomètre
KEYNOTE-775 : Étude contrôlée du traitement en association chez les patientes atteintes d'un CE avancé préalablement traitées par chimiothérapie systémique
L'efficacité du pembrolizumab en association au lenvatinib a été évaluée dans KEYNOTE-775, une étude randomisée, multicentrique, en ouvert, contrôlée versus traitement actif, réalisée chez des patientes atteintes d'un CE avancé préalablement traitées par au moins une chimiothérapie antérieure à base de sels de platine reçue quel que soit le stade, y compris néoadjuvant et adjuvant. Les participantes peuvent avoir reçu jusqu'à 2 thérapies à base de sels de platine au total, à condition que l'une d'entre elles ait été administrée dans le cadre d'un traitement néoadjuvant ou adjuvant. L'étude a exclu les patientes atteintes d'un sarcome de l'endomètre, d'un carcinosarcome, d'une fistule préexistante de Grade ≥ 3, d'une pression artérielle (PA) non contrôlée (> 150/90 mmHg), d'une atteinte ou d'un événement cardiovasculaire significatif au cours des 12 derniers mois, ou des patientes atteintes d'une maladie auto-immune active ou dont l'état clinique nécessitait un traitement immunosuppresseur. La randomisation a été stratifiée selon le statut MMR (dMMR ou pMMR [mismatch repair proficient]) à l'aide d'un test IHC validé. La strate pMMR a ensuite été stratifiée selon le statut de performance ECOG, de la région géographique et des antécédents de radiothérapie pelvienne. Les patientes ont été randomisées (1 : 1) dans l'un des bras de traitement suivants :
· pembrolizumab 200 mg toutes les 3 semaines par voie intraveineuse en association au lenvatinib 20 mg par voie orale, une fois par jour.· au choix de l'investigateur composé soit de doxorubicine 60 mg/m2 toutes les 3 semaines, soit de paclitaxel 80 mg/m2 par semaine, 3 semaines de traitement/1 semaine d'arrêt .
Le traitement par pembrolizumab et lenvatinib a été poursuivi jusqu'à progression de la maladie définie par RECIST v1.1 telle que déterminée en aveugle par une revue centralisée indépendante, toxicité inacceptable, ou, pour pembrolizumab, une durée maximale de 24 mois. L'administration du traitement à l'étude était autorisée au-delà de la progression de la maladie définie par RECIST si l'investigateur considérait qu'il y avait un bénéfice clinique pour la patiente et que le traitement était toléré. Un total de 121/411 (29 %) des patientes traitées par pembrolizumab et lenvatinib ont poursuivi le traitement à l'étude au-delà de la progression de la maladie définie par RECIST. La durée médiane du traitement post-progression était de 2,8 mois. Une évaluation du statut tumoral était réalisée toutes les 8 semaines.
Un total de 827 patientes ont été incluses et randomisées pour recevoir pembrolizumab en association au lenvatinib (n = 411) ou au choix de l'investigateur : doxorubicine (n = 306) ou paclitaxel (n = 110). Les caractéristiques de ces patientes à l'inclusion étaient : âge médian de 65 ans (30 à 86 ans) ; 50 % âgées de 65 ans ou plus ; 61 % de type Caucasien, 21 % de type Asiatique et 4 % de type Noir ; statut de performance ECOG de 0 (59 %) ou 1 (41 %,) et 84 % avec le statut tumoral pMMR et 16 % avec le statut tumoral dMMR. Les sous-types histologiques étaient le carcinome endométrioïde (60 %), séreux (26 %), carcinome à cellules claires (6 %), mixte (5 %) et autre (3 %). Toutes les 827 patientes ont reçu un traitement systémique antérieur pour le CE : 69 % en avaient reçu un, 28 % en avaient reçu deux et 3 % avaient reçu au moins trois traitements systémiques antérieurs. 37 % des patientes n'ont reçu qu'un traitement néoadjuvant ou adjuvant antérieur.
Les critères principaux d'évaluation de l'ef icacité étaient l'OS et la PFS (tels qu'évalués en aveugle par une revue centralisée indépendante utilisant RECIST 1.1). Les critères secondaires d'évaluation de l'ef icacité incluait l'ORR, tels qu'évalués en aveugle par une revue centralisée indépendante utilisant RECIST 1.1. Lors de l'analyse intermédiaire pré-définie avec un suivi médian de 11,4 mois (de 0,3 à 26,9 mois), l'étude a démontré une amélioration statistiquement significative de l'OS et de la PFS. L'analyse finale pré-définie de l'OS avec environ 16 mois de suivi supplémentaire à partir de l'analyse intermédiaire (durée de suivi médiane globale de 14,7 mois [de 0,3 à 43,0 mois]) a été réalisée sans ajustement de multiplicité. Les résultats d'ef icacité par sous-groupes MMR étaient concordants avec les résultats globaux de l'étude. Les résultats de la PFS, de l'ORR et de la durée de réponse lors de l'analyse intermédiaire et les résultats de l'OS lors de l'analyse finale sont résumés dans le tableau 42. Les courbes de Kaplan-Meier pour les analyses finales de l'OS et intermédiaires de la PFS sont présentées dans les figures 36 et 37, respectivement.
Tableau 42 : Résultats d'ef icacité dans KEYNOTE-775
| Critère d'évaluation | Pembrolizumab 200 toutes les3 semaines Lenvatinibn = 411 | Chimiothérapie*n = 416 |
| OS | ||
| Nombre (%) de patientes avec événement | 276 (67 %) | 329 (79 %) |
| Médiane en mois (IC 95 %) | 18,7 (15,6 - 21,3) | 11,9 (10,7 - 13,3) |
| Hazard ratio† (IC 95 %) | 0,65 (0,55 - 0,77) | |
| Valeur de pÞ | < 0,0001 | |
| PFSß | ||
| Nombre (%) de patientes avec événement | 281 (68 %) | 286 (69 %) |
| Médiane en mois (IC 95 %) | 7,2 (5,7 - 7,6) | 3,8 (3,6 - 4,2) |
| Hazard ratio† (IC 95 %) | 0,56 (0,47 - 0,66) | |
| Valeur de p‡ | < 0,0001 | |
| Taux de réponse objectiveß | ||
| ORR§ % (IC 95 %) | 32 % (27 - 37) | 15 % (11 - 18) |
| Réponse complète | 7 % | 3 % |
| Réponse partielle | 25 % | 12 % |
| Valeur de p¶ | < 0,0001 | |
| Durée de réponseß | ||
| Médiane en mois# (intervalle) | 14,4 (1,6+ - 23,7+) | 5,7 (0,0+ - 24,2+) |
† Sur la base du modèle Cox regression stratifié
Þ Valeur de p nominale unilatérale pour l'analyse finale basée sur un test de Log rank stratifié. Lors de l'analyse intermédiaire pré-définie de l'OS avec un suivi médian de 11,4 mois (de 0,3 à 26,9 mois), une supériorité statistiquement significative a été obtenue pour l'OS en comparant l'association de pembrolizumab et lenvatinib à la chimiothérapie (HR : 0,62 ; [IC 95 % : 0,51- 0,75] ; Valeur de p < 0,0001)
ß Lors d'une analyse intermédiaire prédéfinie
‡ Valeur de p unilatérale sur la base du test de Log rank stratifié
§ Réponse : meilleure réponse objective telle que réponse complète ou partielle confirmées
¶ Sur la base de la méthode de Miettinen et Nurminen stratifiée par statut MMR, statut de performance ECOG, région géographique et antécédents de radiothérapie pelvienne
# Sur la base des estimations de Kaplan-Meier
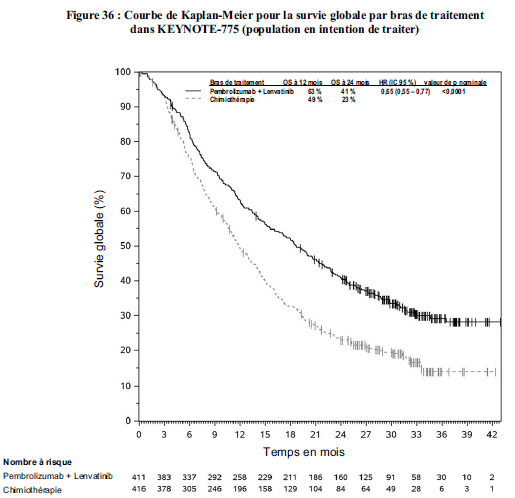
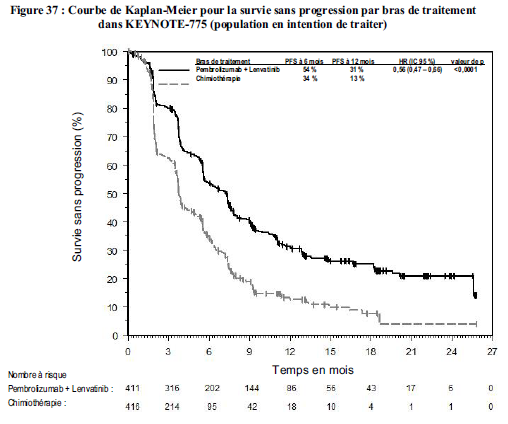
Cancer du col de l'utérus
KEYNOTE-826 : Étude contrôlée du traitement en association chez les patientes atteintes d'un cancer du col de l'utérus persistant, récidivant ou métastatique
L'efficacité du pembrolizumab en association au paclitaxel et cisplatine ou paclitaxel et carboplatine, avec ou sans bevacizumab, a été étudiée dans KEYNOTE-826, une étude multicentrique, randomisée, en double aveugle, contrôlée versus placebo ayant inclus 617 patientes atteintes d'un cancer du col de l'utérus persistant, récidivant, ou métastatique en première ligne n'ayant pas reçu de chimiothérapie sauflorsqu'elle était utilisée simultanément comme agent radiosensibilisant. Les patientes ont été incluses quel que soit le statut d'expression tumorale de PD-L1. Les patientes présentant une maladie auto-immune ayant nécessité un traitement systémique dans les 2 ans précédents le traitement ou un état clinique nécessitant un traitement immunosuppresseur étaient inéligibles. La randomisation a été stratifiée selon le statut métastatique au moment du diagnostic initial, la décision de l'investigateur d'utiliser le bevacizumab, et le statut PD-L1 (CPS < 1 vs. CPS 1 à < 10 vs. CPS ≥ 10). Les patientes ont été randomisées (1 : 1) dans l'un des deux groupes de traitement :
· Groupe de traitement 1 : Pembrolizumab 200 mg plus chimiothérapie avec ou sans bevacizumab· Groupe de traitement 2 : Placebo plus chimiothérapie avec ou sans bevacizumab
L'investigateur a sélectionné l'un des quatre schémas thérapeutiques suivants avant la randomisation :
1. Paclitaxel 175 mg/m2 + cisplatine 50 mg/m22. Paclitaxel 175 mg/m2 + cisplatine 50 mg/m2 + bevacizumab 15 mg/kg
3. Paclitaxel 175 mg/m2 + carboplatine ASC 5 mg/mL/min
4. Paclitaxel 175 mg/m2 + carboplatine ASC 5 mg/mL/min + bevacizumab 15 mg/kg
Tous les médicaments à l'étude ont été administrés par perfusion intraveineuse. Tous les traitements à l'étude ont été administrés le Jour 1 de chaque cycle de traitement de 3 semaines. Le cisplatine pouvait être administré le Jour 2 de chaque cycle de traitement de 3 semaines. L'option d'utiliser le bevacizumab était au choix de l'investigateur avant la randomisation. Le traitement par pembrolizumab a été poursuivi jusqu'à progression de la maladie définie par RECIST v1.1, toxicité inacceptable ou un maximum de 24 mois. L'administration de pembrolizumab était autorisée au-delà de la progression de la maladie définie par RECIST si le patient était cliniquement stable et si l'investigateur considérait qu'il y avait un bénéfice clinique. Une évaluation du statut tumoral était réalisée à la Semaine 9, et ensuite toutes les 9 semaines pendant la première année, puis toutes les 12 semaines par la suite.
Sur les 617 patientes incluses, 548 patientes (89 %) avaient des tumeurs exprimant PD-L1 avec un CPS ≥ 1 sur la base du kit PD-L1 IHC 22C3 pharmDxTM. Parmi ces 548 patientes incluses avec des tumeurs exprimant PD-L1, 273 patientes ont été randomisées pour recevoir pembrolizumab en association à une chimiothérapie avec ou sans bevacizumab, et 275 patientes ont été randomisées pour recevoir un placebo en association à une chimiothérapie avec ou sans bevacizumab. Les caractéristiques à l'inclusion de ces 548 patientes comprenaient : âge médian de 51 ans (intervalle : 22 à 82), 16 % âgés de 65 ans ou plus ; 59 % de type Caucasien, 18 % de type Asiatique et 1 % de type Noir ; 37 % de type Hispanique ou Latino- Américain ; 56 % et 43 % avaient respectivement un statut de performance ECOG de 0 ou 1 ; 63 % ont reçu du bevacizumab comme traitement à l'étude ; 21 % présentaient un adénocarcinome et 5 % présentaient une histologie adénosquameuse ; pour les patientes atteintes d'une maladie persistante ou récidivante avec ou sans métastases à distance, 39 % n'avaient reçu qu'une chimioradiothérapie antérieure et 17 % avaient reçu une chimioradiothérapie antérieure et une chirurgie.
Les critères principaux d'évaluation de l'ef icacité étaient l'OS et la PFS telles qu'évaluées par l'investigateur selon RECIST v1.1. Les critères secondaires d'évaluation de l'ef icacité étaient l'ORR et la durée de réponse, telles qu'évaluées par l'investigateur selon RECIST v1.1. L'étude a démontré des améliorations statistiquement significatives de l'OS et de la PFS chez les patientes randomisées dans le bras pembrolizumab en association à une chimiothérapie avec ou sans bevacizumab par rapport au bras placebo en association à une chimiothérapie avec ou sans bevacizumab lors d'une analyse intermédiaire prédéfinie dans la population globale. La durée médiane de suivi était de 17,2 mois (intervalle : 0,3 à 29,4 mois). Le tableau 43 résume les critères clés d'efficacité chez les patientes dont les tumeurs exprimaient PD-L1 avec un CPS ≥ 1 dans KEYNOTE-826 à partir de l'analyse intermédiaire prédéfinie. Les courbes de Kaplan-Meier pour l'OS et la PFS sont présentées dans les figures 38 et 39.
Tableau 43 : Résultats d'ef icacité dans l'étude KEYNOTE-826 pour les patientes avec une expression de PD-L1 (CPS ≥ 1)
| Critère d'évaluation | Pembrolizumab 200 mg toutes les 3 semainesplus chimiothérapie* avec ou sans bevacizumabn = 273 | Placeboplus chimiothérapie* avec ou sans bevacizumabn = 275 |
| OS | ||
| Nombre (%) de patients avec événement | 118 (43 %) | 154 (56 %) |
| Médiane en mois (IC 95 %) | NA (19,8 ; NA) | 16,3 (14,5 ; 19,4) |
| Hazard ratio† (IC 95 %) | 0,64 (0,50 ; 0,81) | |
| Valeur de p‡ | 0,0001 | |
| PFS | ||
| Nombre (%) de patients avec événement | 157 (58 %) | 198 (72 %) |
| Médiane en mois (IC 95 %) | 10,4 (9,7 ; 12,3) | 8,2 (6,3 ; 8,5) |
| Hazard ratio† (IC 95 %) | 0,62 (0,50 ; 0,77) | |
| Valeur de p§ | < 0,0001 | |
| Taux de réponse objective | ||
| ORR¶ % (IC 95%) | 68 % (62 ; 74) | 50 % (44 ; 56) |
| Réponse complète | 23 % | 13 % |
| Réponse partielle | 45 % | 37 % |
| Durée de réponse | ||
| Médiane en mois (intervalle) | 18,0 (1,3+ ; 24,2+) | 10,4 (1,5+ ; 22,0+) |
| % avec durée ≥ 12 mois# | 56 | 46 |
† Sur la base du modèle Cox proportional hazard stratifié
‡ Sur la base du test de log rank stratifié (par rapport à une limite alpha de 0,00549)
§ Sur la base du test de log rank stratifié (par rapport à une limite alpha de 0,00144)
¶ Réponse : meilleure réponse objective telle que réponse complète ou partielle confirmées
# Sur la base des estimations de Kaplan-Meier NA = non atteint

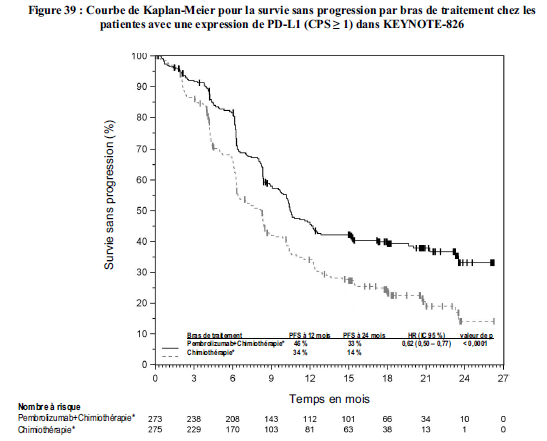
Population âgée
Aucune différence globale en termes de sécurité n'a été observée chez les patients ≥ 75 ans par rapport aux patients plus jeunes recevant le pembrolizumab en monothérapie. Lorsqu'il est administré en association à une chimiothérapie et sur la base de données de sécurité limitées chez les patients ≥ 75 ans, le pembrolizumab a montré une tolérance moindre chez les patients ≥ 75 ans par rapport aux patients plus jeunes. Pour les données d'efficacité chez les patients ≥ 75 ans, veuillez-vous référer à la section appropriée de chaque indication.
Population pédiatrique
Dans l'étude KEYNOTE-051, 161 patients pédiatriques (62 enfants âgés de 9 mois à moins de 12 ans et 99 adolescents âgés de 12 à 17 ans) atteints d'un mélanome avancé ou de tumeurs solides avancées PD-L1 positives, en rechute ou réfractaires, ou d'un lymphome ont reçu pembrolizumab à 2 mg/kg de poids corporel toutes les 3 semaines. Tous les patients ont reçu une médiane de 4 doses de pembrolizumab (1 - 35 doses), avec 138 patients (85,7 %) ayant reçu 2 doses de pembrolizumab ou plus. Les sujets ont été inclus parmi 28 types de tumeurs lors du diagnostic primaire. Les types histologiques de tumeurs les plus fréquents étaient : lymphome de Hodgkin (13,7 %), glioblastome multiforme (9,3 %), neuroblastome (6,2 %), ostéosarcome (6,2 %) et mélanome (5,6 %). Sur les 161 patients, 137 ont été inclus avec des tumeurs solides, 22 avec un lymphome de Hodgkin et 2 avec d'autres lymphomes. Chez les patients avec des tumeurs solides et d'autres lymphomes, l'ORR était de 5,8 %, aucun patient n'avait une réponse complète et 8 patients (5,8 %) avaient une réponse partielle. Dans la population avec un lymphome de Hodgkin (n = 22), chez les patients âgés de 11 à 17 ans, les caractéristiques à l'inclusion étaient : âge médian de 15 ans, 64 % de sexe masculin, 68 % de type caucasien, 77 % avaient un score de performance de Lansky/Karnofsky de 90-100 et 23 % un score de performance de 70-80. Quatre-vingt-six pourcent avaient au moins deux lignes de traitement antérieures et 64 % avaient un stade 3 ou plus élevé. Chez ces patients pédiatriques atteints d'un LHc, l'ORR évalué en aveugle par une revue centralisée indépendante selon les critères de l'IWG de 2007 était de 54,5 %, 1 patient (4,5 %) avait une réponse complète et 11 patients (50,0 %) avaient une réponse partielle, et l'ORR évalué par les critères Lugano de 2014 était de 63,6 %, 4 patients (18,2 %) avaient une réponse complète et 10 patients (45,5 %) avaient une réponse partielle. Les données issues des essais cliniques chez les patients adolescents atteints de mélanome sont très limitées et l'efficacité a été établie par extrapolation à partir des données chez l'adulte. Parmi les 5 adolescents participants atteints de mélanome avancé traités dans KEYNOTE-051, aucun patient n'a eu de réponse complète ou partielle, et 1 patient avait une maladie stable.
L'Agence européenne des médicaments a dif éré l'obligation de soumettre les résultats d'études réalisées avec pembrolizumab dans un ou plusieurs sous-groupes de la population pédiatrique dans le traitement du lymphome de Hodgkin (voir rubrique 4.2 pour les informations concernant l'usage pédiatrique).
La pharmacocinétique de pembrolizumab a été étudiée chez 2 993 patients atteints de mélanome, de CBNPC ou de carcinome métastatiques ou non résécables ayant reçu des doses allant de 1 à 10 mg/kg de poids corporel toutes les 2 semaines, 2 à 10 mg/kg de poids corporel toutes les 3 semaines ou 200 mg toutes les3 semaines.
Absorption
Pembrolizumab est administré par voie intraveineuse, la biodisponibilité est donc immédiate et complète.
Distribution
En cohérence avec une distribution extravasculaire limitée, le volume de distribution de pembrolizumab à l'état d'équilibre est faible (~6,0 L ; coefficient de variation CV : 20 %). Pembrolizumab ne se lie pas aux protéines plasmatiques de manière spécifique, ce qui est prévisible pour un anticorps.
Biotransformation
Pembrolizumab est catabolisé par des voies non-spécifiques, son métabolisme ne contribue pas à sa clairance.
Élimination
Après atteinte de la variation maximale à l'état d'équilibre, la clairance du pembrolizumab est approximativement 23 % plus faible (moyenne géométrique, 195 mL/jour [% CV : 40 %] par rapport à la première dose (252 mL/jour [% CV : 37 %]) ; cette baisse de la clairance avec le temps n'est pas considérée comme cliniquement significative. La valeur de la moyenne géométrique (% CV) pour la demi-vie terminale est de 22 jours (32 %) à l'état d'équilibre.
Linéarité/non-linéarité
L'exposition à pembrolizumab, exprimée par la concentration maximale (Cmax) ou par l'aire sous la courbe (ASC) de la concentration plasmatique en fonction du temps, a augmenté proportionnellement à la dose administrée, dans l'intervalle de doses utilisées pour la mesure de l'ef icacité. Les concentrations de pembrolizumab à l'état d'équilibre ont été atteintes après 16 semaines d'administrations répétées toutes les 3 semaines et le ratio d'accumulation systémique était de 2,1. Les concentrations minimales (Cmin) médianes à l'état d'équilibre étaient approximativement de 22 µg/mL à la dose de 2 mg/kg de poids corporel toutes les 3 semaines et de 29 µg/mL à la dose de 200 mg toutes les 3 semaines. L'aire médiane sous la courbe de la concentration en fonction du temps à l'état d'équilibre sur 3 semaines (ASC0-3semaines) était de 794 µg•jour/mL à la dose de 2 mg/kg de poids corporel toutes les 3 semaines et de 1 053 µg•jour/mL à la dose de 200 mg toutes les 3 semaines.
Suite à l'administration de 200 mg de pembrolizumab toutes les 3 semaines chez des patients atteints de LHc, la Cmin médiane observée à l'état d'équilibre était jusqu'à 40 % plus élevée que celle observée dans d'autres types de tumeur traités avec la même posologie ; cependant la fourchette des concentrations minimales est similaire. Il n'y a pas de dif érence notable de la Cmax médiane entre le LHc et d'autres types de tumeur. Sur la base des données de sécurité disponibles dans le LHc et dans d'autres types de tumeur, ces différences ne sont pas cliniquement significatives.
Populations particulières
Les effets de diverses covariables sur la pharmacocinétique de pembrolizumab ont été évalués par une analyse pharmacocinétique de population. Les facteurs suivants n'ont pas montré d'ef et cliniquement important sur la clairance de pembrolizumab : l'âge (de 15 à 94 ans), le sexe, l'origine ethno-géographique, une insuf isance rénale légère ou modérée, une insuffisance hépatique légère ou modérée et la charge tumorale. La relation entre le poids corporel et la clairance est en faveur de l'utilisation d'une dose fixe ou d'une dose basée sur le poids pour assurer un contrôle adéquat et similaire de l'exposition. L'exposition au pembrolizumab de la population pédiatrique (≥ 3 à 17 ans), avec une posologie basée sur le poids de 2 mg/kg de poids corporel toutes les 3 semaines, est comparable à celle des adultes avec la même dose.
Insuffisance rénale
L'effet de l'insuffisance rénale sur la clairance de pembrolizumab a été évalué par une analyse pharmacocinétique de population chez des patients présentant une insuffisance rénale légère ou modérée par rapport à des patients ayant une fonction rénale normale. Aucune différence cliniquement importante dans laclairance de pembrolizumab n'a été mise en évidence entre les patients présentant une insu f isance rénale légère ou modérée et les patients ayant une fonction rénale normale. Pembrolizumab n'a pas été étudié chez les patients ayant une insuf isance rénale sévère (voir rubrique Posologie et mode d'administration).
Insuffisance hépatique
L'effet de l'insuffisance hépatique sur la clairance de pembrolizumab a été évalué par une analyse pharmacocinétique de population chez des patients présentant une insuffisance hépatique légère et modérée (selon les critères de dysfonction hépatique du US National Cancer Institute) par rapport aux patients ayant une fonction hépatique normale. Aucune différence cliniquement importante dans la clairance depembrolizumab n'a été mise en évidence entre les patients atteints d'insu f isance hépatique légère ou modérée et ceux ayant une fonction hépatique normale. Pembrolizumab n'a pas été étudié chez les patients ayant une insuffisance hépatique sévère (voir rubrique Posologie et mode d'administration).
Pembrolizumab a une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines. Chez certains patients, des étourdissements et de la fatigue ont été rapportés après administration de pembrolizumab (voir rubrique Effets indésirables).
La tolérance de pembrolizumab a été évaluée dans des études de toxicité à doses répétées de 1 et 6 mois chez des singes Cynomolgus ayant reçu des doses intraveineuses de 6, 40 ou 200 mg/kg de poids corporel une fois par semaine dans l'étude de 1 mois et une fois toutes les deux semaines dans celle de 6 mois, suivie d'une période sans traitement de 4 mois. Aucun résultat toxicologique significatif n'a été observé et la dose sans effet toxique bservable (« no observable adverse effect level » [NOAEL]) dans les deux études était ≥ 200 mg/kg de poids corporel, ce qui a conduit à des expositions 19 et 94 fois plus élevées que l'exposition chez l'Homme aux doses de 10 et 2 mg/kg de poids corporel, respectivement. La dose NOAEL correspond à une exposition 74 fois plus élevée qu'à la dose de 200 mg chez l'Homme.
Aucune étude sur la reproduction animale n'a été menée avec pembrolizumab. On estime que la voie anti PD-1/PD-L1 est impliquée dans le maintien de la tolérance au fœtus pendant la grossesse. Il a été montré que le blocage de la signalisation de PD-L1 dans des modèles murins de grossesse induit une modification de la tolérance au fœtus et conduit à une augmentation des pertes fœtales.
Aucune étude de fertilité chez l'animal n'a été conduite avec pembrolizumab. Dans des études de toxicité à doses répétées de 1 et 6 mois chez des singes, aucun effet notable sur les organes reproducteurs mâles et femelles n'a été constaté ; cependant, de nombreux animaux dans ces études n'étaient pas sexuellement matures.
Préparation et administration de la perfusion
· N'agitez pas le flacon.· Laissez revenir le flacon à température ambiante (à 25°C ou en dessous).
· Avant dilution, le flacon de liquide peut être conservé non réfrigéré (températures à 25°C ou en dessous) jusqu'à 24 heures.
· Avant administration, les médicaments à usage parentéral doivent être inspectés visuellement pour détecter la présence éventuelle de particules ou d'un jaunissement. La solution à diluer est une solution limpide à légèrement opalescente, incolore à légèrement jaune. Jetez le flacon si des particules visibles sont observées.
· Retirez le volume nécessaire, jusqu'à 4 mL (100 mg) de solution à diluer et transférez-le dans une poche de perfusion intraveineuse contenant du chlorure de sodium à 9 mg/mL (0,9 %) ou du glucose à 50 mg/mL (5 %) pour préparer une solution diluée avec une concentration finale comprise entre 1 et 10 mg/mL. Chaque flacon contient un excédent de 0,25 mL (contenu total de 4,25 mL par flacon) pour assurer l'extraction de 4 mL de solution à diluer. Mélangez la solution diluée en la renversant doucement.
· D'un point de vue microbiologique, le produit, une fois dilué, doit être utilisé immédiatement. La solution diluée ne doit pas être congelée. Si elle n'est pas utilisée immédiatement, la stabilité chimique et physique de KEYTRUDA en cours d'utilisation a été démontrée pendant 96 heures entre 2°C et 8°C. Cette durée de 96 heures peut inclure jusqu'à 6 heures à température ambiante (à 25°C ou en dessous). En cas de réfrigération, il faut laisser les flacons et/ou les poches de perfusion intraveineuse revenir à température ambiante avant utilisation. Des particules protéiques translucides à blanches peuvent être visibles dans la solution diluée. Administrez la solution pour perfusion par voie intraveineuse sur 30 minutes en utilisant un filtre en ligne ou additionnel, stérile, non pyrogène, à faible taux d'adsorption des protéines de 0,2 à 5 µm.
· Ne pas co-administrer d'autres médicaments par la même ligne de perfusion.
· KEYTRUDA est à usage unique. Jetez tout contenu non utilisé restant dans le flacon.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Médicament nécessitant une surveillance particulière pendant le traitement.
Prescription
réservée aux médecins compétents en maladie du sang.
Prescription réservée aux spécialistes et services CANCEROLOGIE.
Prescription
réservée aux spécialistes et services HEMATOLOGIE.
Prescription réservée aux spécialistes et services ONCOLOGIE MEDICALE.
Réservé à l'usage HOSPITALIER.
Solution à diluer pour perfusion.
Solution limpide à légèrement opalescente, incolore à légèrement jaune, pH 5,2 - 5,8.
Flacon
en verre transparent de type I de 10 mL avec un bouchon gris en
chlorobutyle ou en bromobutyle enrobé et un opercule amovible en
aluminium avec un capuchon flip-off de couleur bleu foncé. Un flacon
contient 4 mL de solution à diluer, contenant 100 mg de pembrolizumab,
Chaque boîte contient un flacon.
Un flacon de 4 mL de solution à diluer contient 100 mg de pembrolizumab.
Chaque mL de solution à diluer contient 25 mg de pembrolizumab.
Pembrolizumab est un anticorps monoclonal humanisé (IgG4 isotype kappa avec altération stabilisatrice de séquence dans la région Fc) anti-PD-1 (programmed cell death-1), produit dans des cellules d'ovaires de hamster chinois par la technique de l'ADN recombinant.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
L-histidine
Chlorhydrate de L-histidine monohydraté
Saccharose
Polysorbate-80 (E433)
Eau pour préparations injectables