
ERELZI 50 mg, solution injectable en stylo prérempli, coffret de 4 stylos préremplis de 1 mL
Dernière révision : 10/10/2024
Taux de TVA : 2.1%
Prix de vente : 472,63 €
Taux remboursement SS : 65%
Base remboursement SS : 472,63 €
Laboratoire exploitant : SANDOZ
Source :

Polyarthrite rhumatoïde
Erelzi en association au méthotrexate est indiqué pour le traitement de la polyarthrite rhumatoïde modérément à sévèrement active de l'adulte en cas de réponse inadéquate aux traitements de fond, y compris le méthotrexate (sauf contre-indication).
Erelzi peut être donné en monothérapie en cas d'intolérance au méthotrexate ou lorsque la poursuite du traitement avec le méthotrexate est inadaptée.
Erelzi est également indiqué dans le traitement de la polyarthrite rhumatoïde sévère, active et évolutive de l'adulte non précédemment traité par le méthotrexate.
Il a été montré que l'étanercept, seul ou en association avec le méthotrexate, ralentit la progression des dommages structuraux articulaires tels que mesurés par la radiographie et améliore les capacités fonctionnelles.
Arthrite juvénile idiopathique
Traitement de la polyarthrite (facteur rhumatoïde positif ou négatif) et de l'oligoarthrite extensive de l'enfant à partir de 2 ans et de l'adolescent en cas de réponse inadéquate ou d'intolérance avérée au méthotrexate.
Traitement de l'arthrite psoriasique de l'adolescent à partir de l'âge de 12 ans en cas de réponse inadéquate ou d'intolérance avérée au méthotrexate.
Traitement de l'arthrite liée à l'enthésite de l'adolescent à partir de l'âge de 12 ans en cas de réponse inadéquate ou d'intolérance avérée au traitement de référence.
Rhumatisme psoriasique
Traitement du rhumatisme psoriasique actif et évolutif de l'adulte en cas de réponse inadéquate au traitement de fond antérieur. Il a été montré que l'étanercept améliore les capacités fonctionnelles chez les patients atteints de rhumatisme psoriasique, et ralentit la progression des dommages structuraux articulaires périphériques tels que mesurés par la radiographie chez les patients ayant des formes polyarticulaires symétriques de la maladie.
Spondyloarthrite axiale
Spondylarthrite ankylosante (SA)
Traitement de la spondylarthrite ankylosante sévère et active de l'adulte en cas de réponse inadéquate au traitement conventionnel.
Spondyloarthrite axiale non radiographique
Traitement de la spondyloarthrite axiale non radiographique sévère de l'adulte avec des signes objectifs d'inflammation, se traduisant par un taux élevé de protéine C réactive (CRP) et/ou des signes visibles à l'imagerie par résonance magnétique (IRM), en cas de réponse inadéquate aux antiinflammatoires non stéroïdiens (AINS).
Psoriasis en plaques
Traitement du psoriasis en plaques modéré
à sévère de l'adulte en cas d'échec, ou de contre-indication, ou d'intolérance
aux autres traitements systémiques y compris la ciclosporine, le méthotrexate
ou la puvathérapie (voir rubrique Propriétés pharmacodynamiques).
Psoriasis en plaques pédiatrique
Traitement du psoriasis en plaques sévère chronique de l'enfant à partir de 6 ans et de l'adolescent en cas de contrôle inadéquat, ou d'intolérance aux autres traitements systémiques ou à la photothérapie.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Septicémie ou risque de septicémie.
Un traitement par Erelzi ne devrait pas être initié chez les patients ayant une infection active y compris les infections chroniques ou localisées.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Infections
Les infections doivent être recherchées chez les patients avant, pendant, et après le traitement par Erelzi, en prenant en compte que la demi-vie d'élimination moyenne d'étanercept est approximativement de 70 heures (entre 7 et 300 heures).
Des infections graves, septicémies, tuberculoses, et infections opportunistes, y compris des infections fongiques invasives, listérioses et légionelloses, ont été rapportées avec l'étanercept (voir rubrique Effets indésirables). Ces infections étaient dues à des bactéries, des mycobactéries, des champignons, des virus et des parasites (y compris des protozoaires). Dans certains cas, des infections fongiques particulières et d'autres infections opportunistes n'ont pas été diagnostiquées, ce qui s'est traduit par un retard d'initiation du traitement approprié et parfois par un décès. Lors de l'évaluation du risque d'infection chez un patient, son exposition à des facteurs de risque spécifiquement associés à certaines infections opportunistes (par exemple, une exposition à des mycoses endémiques) doit être prise en compte.
Une surveillance attentive doit être exercée chez les patients traités par Erelzi développant une nouvelle infection. Le traitement par Erelzi doit être interrompu si le patient développe une infection grave. La sécurité d'emploi et l'efficacité de l'étanercept chez les patients atteints d'infections chroniques n'ont pas été évaluées. Les médecins doivent prescrire Erelzi avec prudence aux patients présentant des antécédents d'infections récurrentes ou chroniques, ou ayant un terrain prédisposant aux infections comme un diabète sévère ou mal équilibré.
Tuberculose
Des cas de tuberculose active comprenant des tuberculoses miliaires et des tuberculoses avec localisation extra-pulmonaire ont été rapportés chez des patients traités par l'étanercept.
Avant de débuter un traitement par Erelzi, une recherche de tuberculose active ou inactive (« latente ») doit être effectuée chez tous les patients. Cette recherche doit comprendre un entretien médical détaillé portant sur les antécédents personnels de tuberculose ou sur d'éventuels contacts antérieurs avec un patient tuberculeux et sur un traitement immunosuppresseur ancien et/ou en cours. Des tests de dépistage appropriés (par exemple, test dermique à la tuberculine et radiographie pulmonaire) doivent être effectués chez tous les patients (conformément aux recommandations locales). Il est recommandé de noter ces tests sur la Carte Patient. Il est rappelé aux prescripteurs que le test dermique à la tuberculine peut s'avérer faussement négatif, en particulier chez un patient sévèrement malade ou immunodéprimé.
Si une tuberculose active est diagnostiquée, le traitement par Erelzi ne doit pas être initié. En cas de diagnostic d'une tuberculose inactive (« latente »), un traitement antituberculeux prophylactique approprié doit être mis en œuvre avant d'initier Erelzi, et en accord avec les recommandations locales. Dans un tel cas, le rapport bénéfice/risque du traitement par Erelzi doit être soigneusement évalué.
Tous les patients devront être informés de la nécessité de consulter un médecin si des signes ou des symptômes évoquant une tuberculose (par exemple, toux persistante, amaigrissement/perte de poids, fébricule) apparaissent pendant ou après le traitement par Erelzi.
Réactivation de l'hépatite B
Une réactivation de l'hépatite B a été rapportée chez des patients précédemment infectés par le virus de l'hépatite B (VHB) et traités par un anti-TNF, y compris l'étanercept. Cela inclut les cas de réactivation de l'hépatite B chez les patients positifs pour les anticorps anti-HBc mais négatifs pour les antigènes HBs. Les patients devront faire l'objet d'un dépistage de l'infection à VHB avant d'initier un traitement par Erelzi. Si les résultats du dépistage sont positifs, il est recommandé de consulter un médecin spécialisé dans le traitement de l'hépatite B. La prudence est de mise lors de l'administration d'Erelzi à des patients présentant des antécédents d'infection par le VHB. Chez ces patients, il faudra surveiller attentivement les signes et les symptômes d'une infection active par le VHB pendant toute la durée du traitement et pendant plusieurs semaines après la fin du traitement. Aucune donnée pertinente pour traiter les patients porteurs de VHB par un traitement antiviral associé à un anti-TNF n'est disponible. Chez les patients qui développent une infection à VHB, le traitement par Erelzi doit être interrompu et un traitement antiviral efficace associé à un traitement symptomatique approprié, doit être instauré.
Aggravation d'hépatite C
Des cas d'aggravation d'hépatite C ont été rapportés chez les patients recevant de l'étanercept. Erelzi doit être utilisé avec prudence chez les patients présentant des antécédents d'hépatite C.
Traitement concomitant avec l'anakinra
L'administration concomitante de l'étanercept et de l'anakinra a été associée à une augmentation du risque d'infections graves et de neutropénies comparativement à l'étanercept administré en monothérapie. Cette association n'a pas démontré un bénéfice clinique supérieur. Par conséquent l'association d'Erelzi et de l'anakinra n'est pas recommandée (voir rubriques Interactions avec d'autres médicaments et autres formes d'interactions et Effets indésirables).
Traitement concomitant avec l'abatacept
L'administration concomitante de l'abatacept et de l'étanercept au cours d'études cliniques a entraîné une augmentation de l'incidence des évènements indésirables graves. Cette association n'a pas démontré de bénéfice clinique supplémentaire. Par conséquent cette association n'est pas recommandée (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Réactions allergiques
Des réactions allergiques associées à l'administration d'étanercept ont été fréquemment rapportées. Ces réactions allergiques ont inclus des cas d'angiœdème et d'urticaire ; des réactions graves se sont produites. En cas de réaction allergique grave ou de réaction anaphylactique, le traitement par Erelzi doit être interrompu immédiatement et un traitement approprié doit être institué.
Immunosuppression
Il est possible que les anti-TNF, y compris Erelzi, altèrent les défenses immunitaires du patient à l'encontre des infections et des tumeurs malignes d'autant que le TNF est un médiateur de l'inflammation et qu'il module la réponse immunitaire des cellules. Dans une étude portant sur 49 patients adultes atteints de polyarthrite rhumatoïde traités par étanercept, il n'a pas été mis en évidence de diminution de l'hypersensibilité de type retardé, de diminution des taux d‘immunoglobulines ou de modification de la numération des populations de cellules effectrices.
Deux patients atteints d'arthrite juvénile idiopathique ont développé une varicelle avec des signes et des symptômes de méningite aseptique suivie d'une guérison sans séquelle. Les patients exposés au virus de la varicelle doivent temporairement arrêter leur traitement par Erelzi et un traitement prophylactique par immunoglobulines spécifiques doit être envisagé.
La sécurité et l'efficacité de l'étanercept chez des patients immunodéprimés n'ont pas été évaluées.
Tumeurs malignes et troubles lymphoprolifératifs
Tumeurs solides et troubles hématopoïétiques (à l'exclusion des cancers cutanés)
Divers cas de tumeurs malignes (cancer du sein, du poumon, lymphome) ont été rapportés au cours de la période post-commercialisation (voir rubrique Effets indésirables).
Dans les phases contrôlées des essais cliniques avec des anti-TNF, plus de cas de lymphomes ont été observés parmi les patients ayant reçu un anti-TNF que chez les patients témoins. Cependant, la survenue était rare et la période de suivi des patients sous placebo était plus courte que celle des patients ayant reçu un traitement par anti-TNF. Après commercialisation, des cas de leucémie ont été rapportés chez des patients traités par anti-TNF. Il existe un risque accru de développer un lymphome ou une leucémie chez les patients atteints de polyarthrite rhumatoïde quand la maladie est ancienne, hautement active et inflammatoire, ce qui complique l'estimation du risque.
Dans l'état actuel des connaissances, la possibilité d'un risque de développer des lymphomes, des leucémies ou d'autres tumeurs malignes solides ou hématopoïétiques chez les patients traités par antiTNF ne peut être écartée. La prudence est de mise lors de l'utilisation d'un traitement par anti-TNF chez des patients présentant des antécédents de tumeur maligne ou lors de la poursuite du traitement chez des patients qui développent une tumeur maligne.
Des tumeurs malignes, dont certaines d'évolution fatale, ont été rapportées après la commercialisation chez des enfants, des adolescents et des jeunes adultes (jusqu'à 22 ans) traités par anti-TNF incluant l'étanercept (initiation du traitement ≤ 18 ans). Environ la moitié des cas était des lymphomes. Les autres cas correspondaient à d'autres types de tumeurs malignes, incluant des tumeurs malignes rares habituellement associées à une immunosuppression. Le risque de développer des tumeurs malignes chez les enfants et les adolescents traités par anti-TNF ne peut être exclu.
Cancers cutanés
Des cas de cancers cutanés mélanomateux et non mélanomateux (CCNM) ont été rapportés chez des patients traités par anti-TNF dont l'étanercept. Des cas de carcinomes à cellules de Merkel ont été rarement rapportés après commercialisation chez des patients traités par l'étanercept. Des examens périodiques de la peau sont recommandés pour tous les patients, particulièrement ceux qui présentent des facteurs de risque de cancer cutané.
En combinant les résultats des essais cliniques contrôlés, un plus grand nombre de cas de CCNM a été observé chez les patients recevant l'étanercept par rapport aux patients témoins, particulièrement chez les patients atteints de psoriasis.
Vaccinations
Les vaccins vivants ne doivent pas être administrés à des patients traités par Erelzi. Aucune donnée n'est disponible sur la transmission infectieuse secondaire à l'administration de vaccins vivants chez des patients traités par l'étanercept. Dans une étude clinique randomisée, contrôlée contre placebo, en double aveugle, menée chez des patients adultes atteints de rhumatisme psoriasique, 184 patients ont également reçu un vaccin pneumococcique polysaccharidique multivalent à la semaine 4. Dans cette étude, la plupart des patients atteints de rhumatisme psoriasique traités par l'étanercept étaient capables d'augmenter la réponse immunitaire des cellules B activées au vaccin pneumococcique polysaccharidique. Cependant, les titres en agrégat étaient modérément bas et quelques patients avaient augmenté leurs titres d'un facteur 2 par rapport aux patients qui n'étaient pas traités par l'étanercept. La signification clinique de ces résultats est inconnue.
Formation d'auto-anticorps
Le traitement par Erelzi est susceptible d'entraîner la formation d'anticorps auto-immuns (voir rubrique Effets indésirables).
Réactions hématologiques
De rares cas de pancytopénie et de très rares cas d'anémie aplasique, dont certains d'évolution fatale, ont été rapportés chez des patients traités par l'étanercept. Une attention particulière doit être portée aux patients traités par Erelzi ayant des antécédents d'atteinte hématologique. Tous les patients et les parents/aidants doivent être informés qu'en cas d'apparition de signes ou de symptômes évoquant une atteinte hématologique ou une infection (tels que fièvre persistante, douleur oropharyngée, ecchymoses, saignement, pâleur) sous Erelzi, ils doivent immédiatement consulter un médecin. Chez ces patients, des examens complémentaires, notamment une numération de la formule sanguine, doivent être pratiqués en urgence ; si une atteinte hématologique est confirmée, le traitement par Erelzi doit être arrêté.
Troubles neurologiques
De rares cas de troubles de démyélinisation du SNC ont été rapportés chez des patients traités par l'étanercept (voir rubrique Effets indésirables). De rares cas de polyneuropathies périphériques démyélinisantes ont également été rapportés (dont le syndrome de Guillain-Barré, la polyneuropathie chronique inflammatoire démyélinisante, la polyneuropathie démyélinisante et la neuropathie motrice multifocale). Bien qu'aucun essai clinique n'ait été réalisé afin d'étudier le traitement par l'étanercept chez des patients atteints de sclérose en plaques, des essais réalisés avec d'autres anti-TNF chez des patients atteints de sclérose en plaques ont mis en évidence une majoration de l'activité de la maladie. Il est recommandé d'évaluer attentivement le rapport bénéfice/risque, avec une évaluation neurologique avant de prescrire Erelzi chez les patients présentant une maladie démyélinisante préexistante ou de survenue récente, ou chez les patients considérés comme ayant un risque accru de développement d'une maladie démyélinisante.
Traitement en association
Au cours d'un essai clinique contrôlé d'une durée de deux ans chez des patients atteints de polyarthrite rhumatoïde, l'association de l'étanercept et du méthotrexate n'a pas révélé de données de sécurité inattendues, et le profil de sécurité de l'étanercept associé au méthotrexate était similaire aux profils rapportés dans les études avec l'étanercept et le méthotrexate utilisés en monothérapie. Des études à long terme visant à évaluer la sécurité de cette association sont actuellement en cours. La sécurité à long terme de l'étanercept en association avec d'autres traitements de fond antirhumatismaux (DMARD) n'a pas été établie.
L'utilisation de l'étanercept en association avec d'autres traitements systémiques ou la photothérapie dans le traitement du psoriasis n'a pas été étudiée.
Insuffisance rénale et hépatique
Sur la base de données de pharmacocinétique (voir rubrique Propriétés pharmacocinétiques) aucun ajustement posologique n'est nécessaire chez les patients présentant une insuffisance rénale ou hépatique ; l'expérience clinique chez de tels patients est limitée.
Insuffisance cardiaque congestive
Les médecins devront utiliser Erelzi avec prudence chez les patients présentant une insuffisance cardiaque congestive (ICC). Des cas d'aggravation d'ICC, avec ou sans facteur favorisant identifiable, chez des patients sous étanercept ont été rapportés après commercialisation. De rares cas (< 0,1 %) d'apparition de novo d'ICC, y compris chez des patients sans maladie cardiovasculaire préexistante connue ont également été rapportés. Certains de ces patients étaient âgés de moins de 50 ans. Deux vastes essais cliniques évaluant l'utilisation de l'étanercept dans le traitement de l'ICC ont pris fin de façon prématurée du fait d'un manque d'efficacité. Bien que non concluantes, les données d'une de ces études suggèrent une tendance possible vers l'aggravation de l'ICC, chez les patients qui recevaient de l'étanercept.
Hépatite alcoolique
Dans une étude de phase II randomisée, contrôlée contre placebo, portant sur 48 patients hospitalisés traités par l'étanercept ou placebo pour une hépatite alcoolique modérée à sévère, l'étanercept n'a pas été efficace et le taux de mortalité des patients traités par l'étanercept était significativement plus élevé après 6 mois. Par conséquent, Erelzi ne doit pas être utilisé dans le traitement de l'hépatite alcoolique. Les médecins devront utiliser Erelzi avec prudence chez les patients présentant également une hépatite alcoolique modérée à sévère.
Granulomatose de Wegener
Un essai contrôlé contre placebo, dans lequel 89 patients adultes étaient traités par l'étanercept ajouté au traitement standard (incluant du cyclophosphamide ou du méthotrexate, et des glucocorticoïdes) pendant une durée médiane de 25 mois, n'a pas démontré que l'étanercept est un traitement efficace dans la granulomatose de Wegener. L'incidence des tumeurs malignes non cutanées de différents types a été significativement plus élevée chez les patients traités par l'étanercept que dans le groupe témoin. Erelzi n'est pas recommandé dans le traitement de la granulomatose de Wegener.
Hypoglycémie chez des patients traités pour un diabète
Des cas d'hypoglycémie ont été rapportés suite à l'initiation de l'étanercept chez des patients qui recevaient un traitement antidiabétique. Ces hypoglycémies ont nécessité une diminution du traitement antidiabétique chez certains de ces patients.
Populations particulières
Personnes âgées
Au cours des études de phase 3 dans la polyarthrite rhumatoïde, le rhumatisme psoriasique et la spondylarthrite ankylosante, aucune différence globale en termes d'événements indésirables, d'événements indésirables graves et d'infections graves n'a été observée chez les patients âgés de 65 ans ou plus recevant l'étanercept par rapport à des patients plus jeunes. Cependant, la prudence s'impose en cas de traitement de personnes âgées et une attention particulière doit être portée vis-à-vis de la survenue d'infections.
Population pédiatrique
Vaccinations
Il est recommandé que les patients pédiatriques aient si possible leurs vaccinations à jour conformément au calendrier de vaccination en vigueur avant d'initier un traitement par Erelzi (voir Vaccinations ci-dessus).
Teneur en sodium
Ce médicament contient moins de 1 mmol (23 mg) de sodium par 25 mg ou 50 mg, c.-à-d. qu'il est essentiellement « sans sodium ».
Résumé du profil de sécurité
Les effets indésirables les plus fréquemment rapportés sont les réactions au site d'injection (telles que douleur, gonflement, démangeaisons, rougeurs et saignement au site d'injection), les infections (telles que les infections des voies aériennes supérieures, les bronchites, les cystites et les infections cutanées), les céphalées, les réactions allergiques, le développement d'auto-anticorps, les démangeaisons et la fièvre.
Des effets indésirables graves ont également été rapportés avec l'étanercept. Les anti-TNF, comme l'étanercept, affectent le système immunitaire et leur utilisation peut affecter les défenses de l'organisme contre l'infection et le cancer. Les infections graves touchent moins de 1 patient sur 100 traités par l'étanercept. Les cas rapportés incluaient des infections fatales, des infections mettant en jeu le pronostic vital et des septicémies. Diverses tumeurs malignes ont également été rapportées avec l'utilisation de l'étanercept, y compris des cancers du sein, du poumon, de la peau et des ganglions lymphatiques (lymphome).
Des effets indésirables hématologiques, neurologiques et auto-immuns graves ont également été rapportés. Ceux-ci incluaient de rares cas de pancytopénie et de très rares cas d'anémie aplasique. Des épisodes de démyélinisation, centrale et périphérique, ont été observés, respectivement rarement et très rarement, au cours de l'utilisation de l'étanercept. De cas rares de lupus, de syndrome lupique et de vascularite ont été observés.
Liste des effets indésirables sous forme de tableau
La liste des effets indésirables ci-dessous est issue de l'expérience des essais cliniques chez l'adulte et des données rapportées depuis la mise sur le marché.
Selon les classes de systèmes d'organe, les effets indésirables sont listés ci-dessous par ordre de fréquence (nombre de patients susceptibles de présenter un effet donné), en utilisant les catégories suivantes : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à <1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000) ; fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
|
Classe de systèmes d'organes |
Très fréquent ≥1/10 |
Fréquent ≥1/100, <1/10 |
Peu fréquent ≥1/1 000, <1/100 |
Rare ≥1/10 000, <1/1 000 |
Très rare <1/10 000 |
Fréquence indéterminée (ne peut être estimée sur la base des données disponibles) |
|
Infections et infestations |
Infections (y compris infection des voies respiratoires supérieures, bronchites, cystites, infection cutanée)* |
|
Infections graves (y compris pneumonie, cellulite, arthrite bactérienne, septicémie et infection parasitaire)* |
Tuberculose, infections opportunistes (incluant infections fongiques invasives, à protozoaires, bactériennes, mycobactériennes atypiques, virales et à légionelle)* |
|
Réactivation de l'hépatite B, listériose |
|
Tumeurs bénignes, malignes et non précisées (incluant kystes et polypes) |
|
|
Cancer cutané non mélanomateux* (voir rubrique Mises en garde spéciales et précautions d'emploi) |
Mélanome malin (voir rubrique Mises en garde spéciales et précautions d'emploi), lymphome, leucémie |
|
Carcinome à cellules de Merkel (voir rubrique Mises en garde spéciales et précautions d'emploi), sarcome de Kaposi |
|
Affections hématologiques et du système lymphatique |
|
|
Thrombopénie, anémie, leucopénie, neutropénie |
Pancytopénie* |
Anémie aplasique* |
Histiocytose hématophagique (syndrome d'activation macrophagique) * |
|
Affections du système immunitaire |
|
Réactions allergiques (voir Affections de la peau et du tissu sous-cutané), formation d'autoanticorps* |
Vascularites (incluant vascularite positive aux anticorps anticytoplasme des polynucléaires neutrophiles) |
Réactions allergiques/ anaphylactiques graves (y compris angiœdème, bronchospasme), sarcoïdose |
|
Aggravation des symptômes de dermatomyosite |
|
Affections du système nerveux |
Céphalées |
|
|
Épisodes de démyélinisation du SNC pouvant évoquer une sclérose en plaques ou un tableau de démyélinisation localisée telle qu'une névrite optique ou une myélite transverse (voir rubrique Mises en garde spéciales et précautions d'emploi), épisodes de démyélinisation périphérique, incluant syndrome de Guillain-Barré, polyneuropathie chronique inflammatoire démyélinisante, polyneuropathie démyélinisante et neuropathie motrice multifocale (voir rubrique Mises en garde spéciales et précautions d'emploi), crise convulsive |
, |
|
|
Affections oculaires |
|
|
Uvéites, sclérites |
|
|
|
|
Affections cardiaques |
|
|
Aggravation d'une insuffisance cardiaque congestive (voir rubrique Mises en garde spéciales et précautions d'emploi) |
Insuffisance cardiaque congestive de novo (voir rubrique Mises en garde spéciales et précautions d'emploi) |
|
|
|
Affections respiratoires, thoraciques et médiastinales |
|
|
|
Pneumopathie interstitielle diffuse (incluant la pneumonie et la fibrose pulmonaire)* |
|
|
|
Affections gastrointestinales |
|
|
Maladie intestinale inflammatoire |
|
|
|
|
Affections hépatobiliaires |
|
|
Élévation des enzymes hépatiques* |
Hépatite autoimmune* |
|
|
|
Affections de la peau et du tissu sous-cutané |
|
Prurit, éruption cutanée |
Angiœdème, psoriasis (y compris première atteinte ou aggravation et atteinte pustuleuse, principalement palmo-plantaire), urticaire, éruption psoriasiforme |
Syndrome de Stevens-Johnson, Vascularite cutanée (incluant la vascularite d'hypersensibilité), érythème polymorphe, |
Nécrolyse épidermique toxique |
|
|
|
|
|
|
réactions lichénoïdes |
|
|
|
Affections musculo- squelettiques et systémiques |
|
|
|
Lupus érythémateux cutané, lupus érythémateux cutané subaigu, syndrome de type lupus |
|
|
|
Affections du rein et des voies urinaires |
|
|
|
Glomérulonéphrite |
|
|
|
Troubles généraux et anomalies au site d'administration |
Réactions au site d'injection (y compris saignement, ecchymoses, érythème, démangeaison s, douleur, gonflement)* |
Fièvre |
|
|
|
|
*Voir Description de certains effets indésirables ci-dessous.
Description de certains effets indésirables
Tumeurs malignes et troubles lymphoprolifératifs
L'apparition de cent vingt-neuf nouvelles tumeurs malignes de différents types a été observée sur un total de 4 114 patients atteints de polyarthrite rhumatoïde traités par l'étanercept dans des essais cliniques jusqu'à 6 ans environ, incluant231 patients traités par l'étanercept associé au méthotrexate dans une étude contrôlée contre comparateur actif d'une durée de 2 ans. Les taux et incidences observés dans ces essais cliniques étaient similaires à ceux attendus dans la population étudiée. Au total, 2 tumeurs malignes ont été rapportées au cours des études cliniques incluant240 patients atteints de rhumatisme psoriasique traités par l'étanercept sur une durée de2 ans environ. Au cours des études cliniques conduites pendant plus de2 ans chez351 patients atteints de spondylarthrite ankylosante, 6tumeurs malignes ont été rapportées chez des patients traités par l'étanercept. Dans un groupe de 2 711 patients atteints de psoriasis en plaques et traités par l'étanercept sur une durée maximale de2,5 ans dans les études en double-aveugle et en ouvert,30tumeurs malignes et43cancers cutanés non mélanomateux ont été rapportés.
Dans un groupe de 7 416 patients traités par l'étanercept au cours des essais cliniques dans la polyarthrite rhumatoïde, le rhumatisme psoriasique, la spondylarthrite ankylosante et le psoriasis,18 lymphomes ont été rapportés.
Divers cas de tumeurs malignes (incluant cancer du sein, du poumon, lymphome) ont été également rapportés au cours de la période post-commercialisation (voir rubrique Mises en garde spéciales et précautions d'emploi).
Réactions au site d'injection
L'incidence des réactions au site de l'injection était significativement plus élevée chez les patients atteints d'affections rhumatismales traités par l'étanercept comparativement au placebo (36 % vs 9 %). Les réactions au site d'injection sont survenues généralement au cours du premier mois de traitement. Leur durée moyenne était d'environ 3 à 5 jours. La majorité des réactions au site d'injection dans les groupes traités par l'étanercept n'a nécessité aucun traitement. La majorité des patients ayant reçu un traitement ont reçu des préparations topiques telles que des corticoïdes ou des antihistaminiques oraux. Par ailleurs, certains patients ont développé des réactions de rappelcaractérisées par l'apparition d'une réaction cutanée au site d'injection le plus récent accompagnée de réactions cutanées aux sites d'injections précédents. Ces réactions étaient généralement transitoires et ne sont pas réapparues lors de la poursuite du traitement.
Au cours des essais contrôlés chez les patients atteints de psoriasis en plaques, environ 13,6 % des patients traités par l'étanercept ont développé des réactions au site d'injection par rapport à 3,4 % des patients sous placebo au cours des 12 premières semaines de traitement.
Infections graves
Au cours des essais contre placebo, aucune augmentation de l'incidence des infections graves (fatales, mettant en jeu le pronostic vital, nécessitant une hospitalisation ou une administration intraveineuse d'antibiotiques) n'a été observée. Des infections graves sont survenues chez 6,3 % des patients atteints de polyarthrite rhumatoïde traités par l'étanercept jusqu'à 48 mois. Ces infections incluaient des abcès (diverses localisations), bactériémie, bronchite, bursite, cellulite, cholécystite, diarrhée, diverticulite, endocardite (suspectée), gastro-entérite, hépatite B, zona, ulcère de la jambe, infection buccale, ostéomyélite, otite, péritonite, pneumonie, pyélonéphrite, septicémie, arthrite septique, sinusite, infection cutanée, ulcère de la peau, infection des voies urinaires, vascularite et plaie infectée. Dans l'étude contrôlée contre comparateur actif d'une durée de 2 ans, dans laquelle les patients étaient traités soit par l'étanercept en monothérapie, soit par le méthotrexate en monothérapie, soit par l'étanercept associé au méthotrexate, les taux d'infections graves étaient similaires parmi les groupes de traitement. Cependant, il ne peut être exclu que l'association del'étanercept au méthotrexate puisse être associée à une augmentation du taux d'infections.
Il n'y a pas eu de différence dans les taux d'infection parmi les patients traités avec l'étanercept et ceux sous placebo pour le psoriasis en plaques dans les essais contrôlés contre placebo d'une durée allant jusqu'à 24 semaines. Les infections graves rapportées chez les patients traités par l'étanercept incluaient cellulite, gastro-entérite, pneumonie, cholécystite, ostéomyélite, gastrite, appendicite, fasciite à streptocoque, myosite, choc septique, diverticulite et abcès. Au cours des essais en double aveugle et en ouvert dans le rhumatisme psoriasique, il a été rapporté un cas d'infection grave (pneumonie).
Des infections graves ou fatales ont été rapportées lors de l'utilisation d'étanercept ; les agents pathogènes identifiés sont des bactéries, des mycobactéries (y compris le bacille de la tuberculose), des virus et des champignons. Certaines sont apparues quelques semaines après le début du traitement par l'étanercept chez des patients ayant des affections sous-jacentes (par exemple diabète, insuffisance cardiaque congestive, antécédents infectieux ou infection chronique) en plus de leur polyarthrite rhumatoïde (voir rubrique4.4). Un traitement par l'étanercept peut augmenter la mortalité chez les patients atteints de septicémie avérée.
Des infections opportunistes ont été rapportées en association avec l'étanercept, notamment des infections fongiques invasives, parasitaires (dont celles à protozoaires), virales (dont zona), bactériennes (dont Listeria et Legionella), et mycobactériennes atypiques. Selon des données combinées des essais cliniques, l'incidence globale des infections opportunistes a été de 0,09 % chez les 15 402 sujets ayant reçu de l'étanercept. Le taux d'événements rapporté à l'exposition a été de 0,06 événement pour 100 patients-année. Environ la moitié des cas d'infections opportunistes rapportés dans le monde après commercialisation étaient des infections fongiques invasives. Les infections fongiques invasives les plus fréquemment rapportées concernaient Candida, Pneumocystis, Aspergillus, et Histoplasma. Plus de la moitié des décès liés à des infections opportunistes était due à des infections fongiques invasives. La majorité des cas de décès concernait des patients atteints de pneumonie à Pneumocystis, d'infection fongique systémique non spécifiée, ou d'aspergillose (voir rubrique Mises en garde spéciales et précautions d'emploi).
Auto-anticorps
Des analyses sanguines à la recherche d'auto-anticorps ont été réalisées à différents moments chez les patients adultes. Parmi les patients atteints de polyarthrite rhumatoïde pour lesquels le taux d'anticorps anti-nucléaires (AAN) a été mesuré, le pourcentage de patients ayant développé de nouveaux AAN positifs (³ 1:40) était plus élevé chez les patients traités par l'étanercept (11 %) que chez les patients sousplacebo (5 %). Le pourcentage de patients ayant développé de nouveaux anticorps anti-ADN double brin positifs était aussi plus élevé par dosage radio-immunologique (15% des patients traités par l'étanercept contre 4 % des patients sous placebo) et par recherche sur Crithidia lucilliae (3 % des patients traités par l'étanercept contre 0 % des patients sous placebo). La proportion de patients traités par l'étanercept ayant développé des anticorps anticardiolipines était augmentée de façon similaire par rapport aux patients sous placebo. L'impact à long terme d'un traitement par l'étanercept sur le développement de maladies auto-immunes est inconnu.
De rares cas de patients (y compris ceux ayant un facteur rhumatoïde positif), ayant développé d'autres auto-anticorps associés à un syndrome lupique ou à des éruptions compatibles sur le plan clinique et après biopsie, avec un lupus cutané subaigu ou un lupus discoïde, ont été rapportés.
Pancytopénie et anémie aplasique
Après commercialisation des cas de pancytopénie et d'anémie aplasique ont été rapportés, dont certains d'issue fatale (voir rubrique Mises en garde spéciales et précautions d'emploi).
Pneumopathie interstitielle diffuse
Au cours des essais cliniques contrôlés menés sur l'étanercept dans toutes les indications, la fréquence (proportion d'incidence) de la pneumopathie interstitielle diffuse chez les patients recevant de l'étanercept sans méthotrexate administré de façon concomitante a été de 0,06 % (rare). Au cours des essais cliniques contrôlés qui autorisaient un traitement concomitant par l'étanercept et le méthotrexate, la fréquence (proportion d'incidence) de la maladie interstitielle du poumon a été de0,47 % (peu fréquent). Après commercialisation, des cas de maladie interstitielle du poumon (incluant la pneumopathie et la fibrose pulmonaire) ont été rapportés, dont certains d'issue fatale.
Traitement concomitant avec l'anakinra
Dans les études où les patients adultes ont reçu un traitement concomitant par l'étanercept et l'anakinra, un taux plus élevé d'infections graves a été observé par rapport à l'étanercept en monothérapie et 2 % des patients (3/139) ont développé une neutropénie (polynucléaires neutrophiles < 1 000/mm3). Tandis qu'il présentait une neutropénie, un patient a développé une cellulite qui a guéri après hospitalisation (voir rubriques Mises en garde spéciales et précautions d'emploi et Interactions avec d'autres médicaments et autres formes d'interactions).
Élévation des enzymes hépatiques
Au cours des périodes en double aveugle des essais cliniques contrôlés menés sur l'étanercept dans toutes les indications, la fréquence (proportion d'incidence) des événements indésirables d'élévation des enzymes hépatiques chez les patients recevant de l'étanercept sans méthotrexate administré de façon concomitante a été de 0,54 % (peu fréquent). Au cours des périodes en double aveugle des essais cliniques contrôlés qui autorisaient un traitement concomitant par l'étanercept et le méthotrexate, la fréquence (proportion d'incidence) des événements indésirables d'élévation des enzymes hépatiques a été de 4,18 % (fréquent).
Hépatite auto-immune
Au cours des essais cliniques contrôlés menés sur l'étanercept dans toutes les indications, la fréquence (proportion d'incidence) de l'hépatite auto-immune chez les patients recevant de l'étanercept sans méthotrexate administré de façon concomitante a été de 0,02 % (rare). Au cours des essais cliniques contrôlés qui autorisaient un traitement concomitant par l'étanercept et le méthotrexate, la fréquence (proportion d'incidence) de l'hépatite auto-immune a été de 0,24 % (peu fréquent).
Population pédiatrique
Effets indésirables chez les patients pédiatriques atteints d'arthrite juvénile idiopathique En général, les événements indésirables chez les patients pédiatriques atteints d'arthrite juvénile idiopathiqueont été similaires en fréquence et en nature à ceux observés chez les patients adultes. Les différences par rapport aux adultes et les autres particularités sont décrites dans les paragraphes suivants.
Les types d'infections rapportés dans les essais cliniques chez des patients atteints d'arthrite juvénile idiopathique âgés de 2 à 18 ans étaient généralement légers à modérés et similaires aux types d'infections communément observés dans les populations pédiatriques ambulatoires. Les événements indésirables sévères rapportés ont été des varicelles avec des signes et symptômes de méningite aseptique suivie d'une guérison sans séquelle (voir également rubrique Mises en garde spéciales et précautions d'emploi), appendicite, gastroentérite, dépression/trouble de la personnalité, ulcère cutané, œsophagite/gastrite, choc septique à streptocoque du groupe A, diabète de type I et infection d'une plaie post-opératoire et du tissu mou.
Dans une étude chez des enfants atteints d'arthrite juvénile idiopathique âgés de 4 à 17 ans, 43 des 69 enfants (62 %) ont présenté une infection en recevant de l'étanercept pendant les 3 mois de l'étude (partie 1 en ouvert) et la fréquence ainsi que la sévérité des infections étaient similaires chez les 58 patients ayant poursuivi l'étude d'extension en ouvert pendant 12 mois. Les types et la proportion des événements indésirables chez les patients atteints d'arthrite juvénile idiopathique étaient similaires à ceux observés dans les essais cliniques de l'étanercept chez les patients adultes atteints de polyarthrite rhumatoïde et étaient en majorité d'intensité légère. Plusieurs événements indésirables ont été rapportés plus fréquemment chez les 69 patients atteints d'arthrite juvénile idiopathique ayant reçu de l'étanercept pendant 3 mois par rapport aux 349 patients adultes atteints de polyarthrite rhumatoïde. Il s'agissait de céphalées (19 % des patients, 1,7 événement par patient-année), nausées (9 %, 1,0 événement par patient-année), douleurs abdominales (19 %, 0,74 événement par patient-année), et vomissements (13 %, 0,74 événement par patient-année).
Quatre cas de syndrome d'activation macrophagique ont été rapportés au cours des essais cliniques dans l'arthrite juvénile idiopathique.
Effets indésirables chez les patients pédiatriques atteints de psoriasis en plaques
Au cours d'une étude sur 48 semaines réalisée chez 211 enfants âgés de 4 à 17 ans et atteints de psoriasis en plaques pédiatrique, les évènements indésirables rapportés ont été similaires à ceux observés dans les études antérieures réalisées chez des adultes atteints de psoriasis en plaques.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir .
- Les infections doivent être
recherchées chez les patients AVANT, PENDANT, et APRES le traitement.
Une recherche de tuberculose active ou inactive (« latente ») doit être
effectuée chez tous les patients (des tests de dépistage appropriés,
par exemple un test dermique à la tuberculine et une radiographie
pulmonaire, devront être effectués chez tous les patients). Il est
recommandé de noter ces examens sur la Carte de Surveillance du patient.
- Des examens périodiques de la peau sont recommandés pour tous les patients, particulièrement ceux qui ont un facteur de risque de cancer cutané.
- VACCINATIONS : il est recommandé que les enfants aient leurs vaccinations à jour conformément au calendrier de vaccination en vigueur avant d'initier un traitement par etanercept.
- Les nourrissons exposés in utero pourraient avoir un risque accru d'infection. Il est déconseillé d'administrer des vaccins vivants à des nourrissons jusqu'à 16 semaines après la dernière dose d'etanercept reçue par la mère pendant la grossesse.
- Des examens périodiques de la peau sont recommandés pour tous les patients, particulièrement ceux qui ont un facteur de risque de cancer cutané.
- VACCINATIONS : il est recommandé que les enfants aient leurs vaccinations à jour conformément au calendrier de vaccination en vigueur avant d'initier un traitement par etanercept.
- Les nourrissons exposés in utero pourraient avoir un risque accru d'infection. Il est déconseillé d'administrer des vaccins vivants à des nourrissons jusqu'à 16 semaines après la dernière dose d'etanercept reçue par la mère pendant la grossesse.
CONTACTER LE MEDECIN IMMEDIATEMENT OU ALLER AU SERVICE DES URGENCES DE VOTRE HOPITAL le plus proche en cas de :
- Troubles de la déglutition ou de la respiration.
- Gonflement de la face, de la gorge, des mains ou des pieds.
- Sentiment de nervosité ou d'anxiété, sensations pulsatiles ou élancements, ou rougeur soudaine de la peau et/ou sensation de chaleur.
- Eruption cutanée sévère, démangeaison, ou urticaire (zones épaissies de peau rouge ou pâle qui souvent démangent).
- Signes d'infections graves, comme une fièvre élevée qui peut être accompagnée de toux, essoufflement, frissons, faiblesse, ou une zone chaude, rouge, douloureuse, irritée sur la peau ou les articulations.
- Signes de troubles sanguins, comme des saignements, ecchymoses (bleus), ou pâleur.
- Signes de troubles du système nerveux, comme des engourdissements ou des fourmillements, troubles de la vision, douleurs oculaires, ou apparition de faiblesse dans les bras ou les jambes.
- Signes d'aggravation d'une insuffisance cardiaque, comme de la fatigue ou de l'essoufflement à l'activité, gonflement des chevilles, sensation d'épaisseur dans le cou ou l'abdomen, essoufflement nocturne ou toux, coloration bleuâtre des ongles ou autour des lèvres.
CONTACTER LE MEDECIN si des signes ou des symptômes évoquant une tuberculose (par exemple, toux persistante, amaigrissement/perte de poids, fébricule) apparaissent pendant ou après le traitement.
FEMME en AGE de PROCREER : utiliser une méthode de contraception pendant toute la durée du traitement et jusqu'à trois semaines après l'arrêt du traitement.
Femmes en âge de procréer
Les femmes en âge de procréer doivent envisager d'utiliser une contraception appropriée pour éviter une grossesse pendant un traitement par Erelzi et jusqu'à trois semaines après l'arrêt du traitement.
Grossesse
Des études de toxicité sur le développement réalisées chez le rat et le lapin n'ont pas mis en évidence de dommage pour le fœtus ou pour le rat nouveau-né qui serait dû à l'étanercept. Les effets de l'étanercept sur l'issue des grossesses ont été étudiés au cours de deux études de cohortes observationnelles. Un taux plus élevé des malformations congénitales majeures a été observé dans le cadre d'une étude observationnelle comparant les grossesses exposées à l'etanercept au cours du premier trimestre (n = 370) aux grossesses non exposées à l'étanercept ou à d'autres anti-TNF (n = 164) (rapport des cotes ajusté de 2,4 ; IC à 95 % : 1,0-5,5). Les types de malformations congénitales majeures correspondaient à ceux les plus fréquemment rencontrés dans la population générale et aucun profil particulier d'anomalie n'a été identifié. Il n'a été observé aucune modification de la fréquence des avortements spontanés, de la mortinatalité, ou des malformations mineures. Dans le cadre d'une autre étude observationnelle du registre menée dans plusieurs pays et comparant le risque d'effets indésirables à l'issue de la grossesse chez les femmes exposées à l'étanercept au cours des 90 premiers jours de grossesse (n = 425) à celles exposées à des médicaments non biologiques (n = 3 497), il n'a été observé aucun risque accru de malformations congénitales majeures (rapport des cotes [odds ratio, OR] non ajusté de 1,22 ; IC à 95 % : 0,79-1,90 ; OR ajusté de 0,96 ; IC à 95 % : 0,58-1,60 après ajustement pour le pays, la maladie maternelle, la parité, l'âge maternel et le tabagisme au début de la grossesse). De plus, cette étude n'a révélé aucun risque accru de malformations congénitales mineures, de naissance prématurée, de mortinatalité ou d'infections au cours de la première année de vie des nourrissons nés de femmes exposées à l'étanercept pendant leur grossesse. Erelzi ne doit être utilisé durant la grossesse qu'en cas de réelle nécessité.
L'étanercept traverse le placenta et a été détecté dans le sérum de nourrissons dont la mère avait été traitée par l'étanercept pendant la grossesse. Les conséquences cliniques ne sont pas connues, mais les nourrissons pourraient être exposés à un risque accru d'infection. Il n'est généralement pas recommandé d'administrer des vaccins vivants à des nourrissons jusqu'à 16 semaines après la dernière dose d'Erelzi reçue par la mère.
Allaitement
Chez les rates allaitantes, après administration sous-cutanée, l'etanercept a été excrété dans le lait et détecté dans le sérum des nouveau-nés. Des informations limitées issues de la littérature publiée indiquent que l'etanercept a été détecté à de faibles taux dans le lait maternel. L'utilisation de l'etanercept pendant l'allaitement pourrait être envisagée en tenant compte du bénéfice de l'allaitement pour l'enfant et du bénéfice du traitement pour la femme.
Bien que l'on s'attende à ce que l'exposition systémique chez un nourrisson allaité soit faible en raison de la dégradation importante de l'etanercept dans le tractus gastro-intestinal, les données disponibles concernant l'exposition systémique chez le nourrisson allaité sont limitées. Par conséquent, l'administration de vaccins vivants (par exemple, BCG) à un nourrisson allaité lorsque la mère reçoit de l'etanercept pourrait être envisagée 16 semaines après l'arrêt de l'allaitement (ou à un moment antérieur si les taux sériques d'etanercept chez le nourrisson sont indétectables).
Fertilité
Il n'existe pas de données précliniques disponibles sur la toxicité péri- et postnatale de l'étanercept, ni sur les effets de l'étanercept sur la fertilité et la fonction reproductrice générale.
Traitement concomitant avec l'anakinra
Les patients adultes traités par l'étanercept et l'anakinra ont présenté un taux plus élevé d'infections graves par rapport aux patients traités soit par l'étanercept en monothérapie, soit par l'anakinra en monothérapie (données historiques).
En outre, au cours d'un essai contrôlé contre placebo, en double aveugle, mené chez des patients adultes recevant un traitement de fond par le méthotrexate, les patients traités par l'étanercept et l'anakinra ont présenté un taux plus élevé d'infections graves (7 %) et de neutropénies que les patients traités par l'étanercept(voir rubriques Mises en garde spéciales et précautions d'emploi et Effets indésirables). L'association de l'étanercept et de l'anakinra n'a pas démontré de bénéfice clinique supérieur et n'est par conséquent pas recommandée.
Traitement concomitant avec l'abatacept
L'administration concomitante de l'abatacept et de l'étanercept au cours d'études cliniques a entraîné une augmentation de l'incidence des événements indésirables graves. Cette association n'a pas démontré de bénéfice clinique supplémentaire. Par conséquent cette utilisation n'est pas recommandée (voir rubrique Mises en garde spéciales et précautions d'emploi).
Traitement concomitant avec la sulfasalazine
Au cours d'une étude clinique menée chez des patients adultes traités par des doses stables de sulfasalazine et chez lesquels de l'étanercept était ajouté, les patients du groupe recevant cette association ont présenté une diminution significative du nombre moyen de globules blancs, par rapport aux groupes traités par l'étanercept ou la sulfasalazine en monothérapie. La signification clinique de cette interaction est inconnue.
Les médecins doivent faire preuve de prudence lorsqu'ils envisagent un traitement en association avec la sulfasalazine.
Absence d'interactions
Au cours des essais cliniques, aucune interaction n'a été observée lorsque l'étanercept était administré avec des glucocorticoïdes, des salicylés (sauf la sulfasalazine), des anti-inflammatoires non stéroïdiens (AINS), des analgésiques ou le méthotrexate. Voir rubrique Mises en garde spéciales et précautions d'emploi pour les recommandations sur la vaccination.
Aucune interaction pharmacocinétique médicamenteuse cliniquement significative n'a été observée au cours des études avec le méthotrexate, la digoxine ou la warfarine.
Le traitement par Erelzi doit être initié et surveillé par un médecin spécialiste ayant l'expérience du diagnostic et du traitement de la polyarthrite rhumatoïde, de l'arthrite juvénile idiopathique, du rhumatisme psoriasique, de la spondylarthrite ankylosante, de la spondyloarthrite axiale non radiographique, du psoriasis en plaques ou du psoriasis en plaques pédiatrique. La Carte Patient devra être remise aux patients traités par Erelzi.
Erelzi est disponible en dosages de 25 mg et de 50 mg.
Posologie
Polyarthrite rhumatoïde
La dose recommandée d'étanercept est de 25 mg administrée deux fois par semaine. Toutefois, l'efficacité et la sécurité d'emploi d'une administration de 50 mg une fois par semaine ont été démontrées (voir rubrique Propriétés pharmacodynamiques).
Rhumatisme psoriasique, spondylarthrite ankylosante et spondyloarthrite axiale non radiographique La dose recommandée est de 25 mg d'étanercept administrée deux fois par semaine ou de 50 mg administrée une fois par semaine.
Pour toutes les indications ci-dessus, les données disponibles laissent supposer qu'une réponse clinique est habituellement obtenue en 12 semaines de traitement. La poursuite du traitement devra être soigneusement reconsidérée chez un patient n'ayant pas répondu dans ces délais.
Psoriasis en plaques
La dose recommandée d'étanercept est de 25 mg administrée deux fois par semaine ou de 50 mg administrée une fois par semaine. Toutefois, une administration de 50 mg deux fois par semaine peut être utilisée jusqu'à 12 semaines, suivie, si nécessaire, par l'administration d'une dose de 25 mg deux fois par semaine ou de 50 mg une fois par semaine. Le traitement par l'étanercept doit être poursuivi jusqu'à l'obtention de la rémission, au maximum jusqu'à 24 semaines. Un traitement continu au-delà de 24 semaines peut être approprié pour certains patients adultes (voir rubrique Propriétés pharmacodynamiques). Le traitement par l'étanercept doit être interrompu chez les patients ne présentant pas de réponse après 12 semaines de traitement. Si la reprise du traitement par l'étanercept est indiquée, le même schéma de durée de traitement doit être suivi. La dose doit être de 25 mg administrée deux fois par semaine ou 50 mg une fois par semaine.
Populations particulières
Insuffisants rénaux et hépatiques
Aucun ajustement posologique n'est nécessaire.
Personnes âgées
Aucun ajustement posologique n'est nécessaire. La posologie et l'administration sont identiques à celles de l'adulte âgé de 18 à 64 ans.
Population pédiatrique
Erelzi est uniquement disponible en seringue préremplie de 25 mg ainsi qu'en seringue préremplie et en stylo prérempli de 50 mg. Il n'est donc pas possible d'administrer Erelzi aux patients pédiatriques ayant besoin d'une dose inférieure à la dose complète de 25 mg ou 50 mg. Les patients pédiatriques ayant besoin d'une dose autre que la dose complète de 25 mg ou 50 mg ne doivent pas recevoir Erelzi. Si une dose différente est requise, d'autres produits à base de l'étanercept permettant d'obtenir cette dose doivent être utilisés.
La dose d'étanercept dépend du poids des patients pédiatriques. Les patients pesant moins de 62,5 kg doivent recevoir une dose exacte calculée en mg/kg en utilisant les présentations poudre et solvant pour solution injectable ou la présentation poudre pour solution injectable (voir ci-dessous pour les doses en fonction des indications). Les patients pesant 62,5 kg ou plus peuvent recevoir une dose fixe en utilisant la seringue préremplie ou le stylo prérempli.
La sécurité et l'efficacité d'Erelzi chez les enfants âgés de moins de 2 ans n'ont pas été établies. Aucune donnée n'est disponible.
Arthrite juvénile idiopathique
La dose recommandée est de 0,4 mg/kg (au maximum 25 mg par injection), administrée deux fois par semaine en injection sous-cutanée, avec un intervalle de 3-4 jours entre deux injections ou de 0,8 mg/kg (au maximum 50 mg par injection) administrée une fois par semaine. L'arrêt du traitement doit être envisagé chez les patients non répondeurs après 4 mois.
Le flacon dosé à 10 mg est plus approprié à l'administration aux enfants souffrant d'AJI ayant un poids inférieur à 25 kg.
Aucun essai clinique n'a été réalisé chez les enfants âgés de 2 à 3 ans. Des données limitées de sécurité provenant d'un registre de patients suggèrent cependant que le profil de sécurité chez les enfants âgés de 2 à 3 ans est similaire à celui des adultes et des enfants âgés de 4 ans et plus, à une dose de 0,8 mg/kg par voie sous-cutanée chaque semaine (voir rubrique Propriétés pharmacodynamiques).
Il n'y a généralement pas lieu d'utiliser l'étanercept chez les enfants âgés de moins de 2 ans dans l'indication arthrite juvénile idiopathique.
Psoriasis en plaques pédiatrique (6 ans et plus)
La dose recommandée est de 0,8 mg/kg (au maximum 50 mg par injection) une fois par semaine jusqu'à 24 semaines. Le traitement doit être interrompu chez les patients ne présentant pas de réponse après 12 semaines de traitement.
Si la reprise du traitement par l'étanercept est indiquée, le schéma de durée du traitement décrit cidessus doit être suivi. La dose doit être de 0,8 mg/kg (au maximum 50 mg par injection) une fois par semaine.
Il n'y a généralement pas lieu d'utiliser l'étanercept chez les enfants âgés de moins de 6 ans dans l'indication psoriasis en plaques.
Mode d'administration
Erelzi est administré par injection sous-cutanée (voir rubrique Précautions particulières d'élimination et de manipulation).
Des instructions complètes pour l'administration sont données dans la notice, à la rubrique 7 « Instructions pour l'utilisation de la seringue préremplie d'Erelzi » ou « Instructions pour l'utilisation du stylo prérempli d'Erelzi ».
Durée de conservation :
3 ans
Précautions particulières de conservation :
A conserver au réfrigérateur (entre 2 C et 8 C).
Ne pas congeler.
Conserver les stylos préremplis dans l'emballage extérieur, à l'abri de la lumière.
Après avoir pris une seringue du réfrigérateur, attendez environ 15-30 minutes afin que la solution d'Erelzi atteigne la température ambiante. Ne pas chauffer d'une quelconque façon. L'utilisation immédiate est alors recommandée.
Erelzi peut être conservé à une température ne dépassant pas 25 °C pendant une durée maximum de 4 semaines, non renouvelable ; après quoi il ne doit pas être mis à nouveau au réfrigérateur. Erelzi doit être éliminé s'il n'est pas utilisé dans les quatre semaines suivant le retrait du réfrigérateur.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Aucune dose limite toxique n'a été observée durant les essais cliniques menés chez les patients atteints de polyarthrite rhumatoïde. La dose la plus élevée ayant été évaluée était une dose de charge intraveineuse de 32 mg/m2, suivie par des doses sous-cutanées de 16 mg/m2 administrées deux fois par semaine. Un patient atteint de polyarthrite rhumatoïde s'est auto-administré par erreur 62 mg d'étanercept par voie sous-cutanée deux fois par semaine pendant trois semaines sans présenter d'effets indésirables. Il n'existe aucun antidote connu à l'étanercept.
Classe pharmacothérapeutique : Immunosuppresseurs, inhibiteurs du facteur nécrosant des tumeurs alpha (TNFα), Code ATC : L04AB01
Erelzi est un médicament biosimilaire. Des informations détaillées sont disponibles sur le site internet de l'Agence européenne des médicaments https://www.ema.europa.eu.
Le facteur nécrosant des tumeurs (TNF) est une cytokine dominante dans le processus inflammatoire de la polyarthrite rhumatoïde. Des taux élevés de TNF sont également retrouvés dans les membranes synoviales et les plaques de psoriasis des patients atteints de rhumatisme psoriasique, et dans le sérum et le tissu synovial des patients atteints de spondylarthrite ankylosante. Dans le psoriasis en plaques, l'infiltration par les cellules inflammatoires, y compris les cellules T, conduit à une augmentation des taux de TNF dans les lésions psoriasiques, comparativement aux taux observés au niveau des zones non atteintes de la peau. L'étanercept est un inhibiteur compétitif de la liaison du TNF à ses récepteurs de surface inhibant ainsi l'activité biologique du TNF. Le TNF et la lymphotoxine sont des cytokines pro-inflammatoires qui lient deux récepteurs distincts à la surface des cellules : les récepteurs du facteur nécrosant des tumeurs (TNFR) de 55-kilodaltons (p55) et de 75-kilodaltons (p75). Ces deux TNFR existent naturellement sous des formes membranaires et solubles. On pense que les TNFR solubles régulent l'activité biologique du TNF.
Le TNF et la lymphotoxine existent principalement sous forme d'homotrimères, leur activité biologique étant dépendante de la réticulation des TNFR à la surface des cellules. Les récepteurs dimères solubles tels qu'étanercept présentent une affinité plus marquée pour le TNF que les récepteurs monomères et sont des inhibiteurs compétitifs beaucoup plus puissants de la liaison du TNF à ses récepteurs cellulaires. En outre, l'utilisation d'une région Fc d'immunoglobuline en tant qu'élément de fusion dans la construction d'un récepteur dimère confère à la molécule une demi-vie plasmatique plus longue.
Mécanisme d'action
La majorité des atteintes articulaires de la polyarthrite rhumatoïde et de la spondylarthrite ankylosante, et des atteintes cutanées du psoriasis en plaques est médiée par des molécules pro-inflammatoires qui appartiennent à un réseau contrôlé par le TNF. Le mécanisme d'action supposé d'étanercept consiste en une inhibition compétitive de la liaison du TNF aux TNFR de la surface cellulaire : les réponses cellulaires médiées par le TNF sont bloquées en rendant le TNF biologiquement inactif. Étanercept pourrait également moduler les réponses biologiques contrôlées par d'autres molécules agissant en aval (par exemple : cytokines, adhésines ou protéinases) dont l'activité est induite ou régulée par le
TNF.
Efficacité et sécurité cliniques
Cette rubrique présente les données issues de quatre études contrôlées randomisées chez l'adulte atteint de polyarthrite rhumatoïde, d'une étude chez l'adulte atteint de rhumatisme psoriasique, d'une étude chez l'adulte atteint de spondylarthrite ankylosante, de deux études chez l'adulte atteint de spondyloarthrite axiale non radiographique, de quatre études chez l'adulte atteint de psoriasis en plaques, de trois études dans l'arthrite juvénile idiopathique et d'une étude chez les patients pédiatriques atteints de psoriasis en plaques.
Patients adultes atteints de polyarthrite rhumatoïde
L'efficacité de l'étanercept a été évaluée au cours d'une étude randomisée, en double aveugle, contrôlée contre placebo. L'étude a évalué 234 patients adultes, présentant une polyarthrite rhumatoïde active, ne répondant pas à au moins un, et au plus quatre traitements de fond antirhumatismaux (DMARD). Des doses de 10 mg ou 25 mg d'étanercept ou du placebo ont été administrées par voie sous-cutanée deux fois par semaine pendant 6 mois consécutifs. Les résultats de cet essai contrôlé ont été exprimés en pourcentage d'amélioration de la polyarthrite rhumatoïde, à l'aide des critères de réponse de l'American College of Rheumatology (ACR).
Les réponses ACR 20 et ACR 50 étaient supérieures chez les patients traités par l'étanercept par rapport au placebo à 3 et 6 mois (ACR 20 : étanercept 62 % et 59 %, placebo 23 % et 11 % à 3 et 6 mois, respectivement ; ACR 50 : étanercept 41 % et 40 %, placebo 8 % et 5 % à 3 et 6 mois, respectivement ; p < 0,01 étanercept vs placebo à tous les temps d'évaluation pour les réponses ACR 20 et ACR 50).
Environ 15 % des sujets recevant l'étanercept ont obtenu une réponse ACR 70 à 3 mois et à 6 mois, comparativement à moins de 5 % des sujets du bras placebo. Parmi les patients recevant l'étanercept, les réponses cliniques ont généralement débuté 1 à 2 semaines après l'initiation du traitement, et ont été quasiment toujours obtenues dans les 3 mois. Une réponse dose-dépendante a été observée ; les résultats avec 10 mg étaient intermédiaires entre le placebo et 25 mg. L'étanercept était significativement supérieur au placebo sur tous les items des critères ACR, ainsi que sur les autres mesures d'activité de la polyarthrite rhumatoïde non compris dans ces critères de réponse ACR, comme la durée de la raideur matinale. L'échelle HAQ (Health Assessment Questionnaire), incluant le handicap, l'activité, l'état mental, l'état général, l'état des fonctions articulaires, a été évaluée tous les 3 mois pendant l'essai. Tous les sous-domaines de l'échelle HAQ ont été améliorés chez les patients traités par l'étanercept par rapport aux témoins à 3 et 6 mois.
Après l'arrêt de l'étanercept, les symptômes d'arthrite sont généralement réapparus au cours du mois suivant. Selon les résultats des études en ouvert, la reprise du traitement par l'étanercept après des arrêts allant jusqu'à 24 mois, a entraîné la même amplitude de réponse que chez les patients recevant l'étanercept sans interruption de traitement. Des réponses stables et durables ont été observées chez des patients recevant l'étanercept sans interruption jusqu'à 10 ans dans les essais thérapeutiques d'extension en ouvert.
L'efficacité de l'étanercept a été comparée avec le méthotrexate au cours d'une étude randomisée, contrôlée contre comparateur actif avec des examens radiographiques réalisés en aveugle comme critère d'évaluation principal, chez 632 patients adultes présentant une polyarthrite rhumatoïde active (de durée < 3 ans) qui n'avaient jamais reçu de traitement par méthotrexate. Des doses de 10 mg ou de 25 mg d'étanercept ont été administrées par voie sous-cutanée deux fois par semaine jusqu'à 24 mois. Les doses de méthotrexate ont été augmentées de 7,5 mg/semaine à 20 mg/semaine maximum au cours des 8 premières semaines de l'essai et maintenues jusqu'à 24 mois. Avec l'étanercept 25 mg, l'amélioration clinique, y compris le délai d'action sous 2 semaines, a été similaire à celle observée lors des essais précédents, et s'est maintenue jusqu'à 24 mois. À l'inclusion, les patients avaient un degré d'invalidité modéré, avec des scores moyens de HAQ de 1,4 à 1,5. Le traitement par l'étanercept à 25 mg a entraîné une amélioration importante à 12 mois, avec environ 44 % de patients obtenant un score de HAQ normal (moins de 0,5). Ce bénéfice a été maintenu la deuxième année de cette étude.
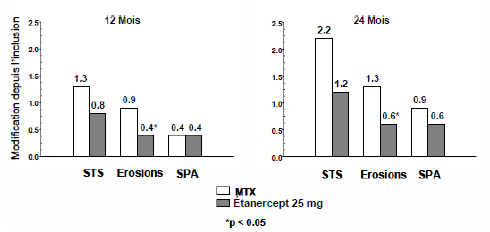
Dans cette étude, les dommages structuraux articulaires ont été évalués radiographiquement et exprimés en modification du Score total de Sharp (STS) et de ses composants ; le score d'érosion et le Score de pincement articulaire (SPA). Les radiographies des mains/poignets et pieds ont été lues à l'inclusion puis à 6, 12 et 24 mois. La dose de 10 mg d'étanercept a eu constamment moins d'effet sur les dommages structuraux que la dose de 25 mg. L'étanercept à 25 mg a été significativement supérieur au méthotrexate pour les scores d'érosion, à la fois à 12 et 24 mois. Les différences entre le groupe méthotrexate et le groupe étanercept à 25 mg pour le STS et le SPA n'étaient pas statistiquement significatives. Les résultats sont présentés dans la figure ci-dessous.
Progression radiographique : comparaison de l'étanercept vs méthotrexate chez des patients ayant une polyarthrite rhumatoïde d'ancienneté inférieure à 3 ans
Dans une autre étude contrôlée contre comparateur actif, en double aveugle, randomisée, l'efficacité clinique, la sécurité, et l'évolution radiographique chez des patients atteints de PR traités par étanercept en monothérapie (25 mg deux fois par semaine), ou méthotrexate en monothérapie (7,5 à 20 mg par semaine, dose médiane 20 mg) ou étanercept associé au méthotrexate débutés simultanément, ont été comparées chez 682 patients adultes ayant une polyarthrite rhumatoïde active d'ancienneté de 6 mois à 20 ans (médiane 5 ans) et qui avaient eu une réponse insuffisante à au moins 1 traitement de fond antirhumatismal (DMARD) autre que le méthotrexate.
Les patients traités par l'étanercept associé au méthotrexate avaient des réponses ACR 20, ACR 50 et
ACR 70 ainsi qu'une amélioration des scores DAS et HAQ significativement plus élevées à la fois à 24 et 52 semaines, comparativement aux patients de chacun des groupes en monothérapie (résultats présentés dans le tableau ci-dessous). Des avantages significatifs avec l'étanercept associé au méthotrexate comparativement à l'étanercept en monothérapie et au méthotrexate en monothérapie ont aussi été observés après 24 mois.
Résultats d'efficacité clinique à 12 mois : comparaison d'étanercept vs méthotrexate vs étanercept associé au méthotrexate chez des patients ayant une PR d'ancienneté de 6 mois à 20 ans
|
Critère d'évaluation |
|
Méthotrexate (n = 228) |
Étanercept (n = 223) |
Étanercept + Méthotrexate (n = 231) |
|
Réponses ACRa |
|
|
|
|
|
|
ACR 20 |
58,8 % |
65,5 % |
74,5 % †, |
|
|
ACR 50 |
36,4 % |
43,0 % |
63,2 % †, |
|
|
ACR 70 |
16,7 % |
22,0 % |
39,8 % †, |
|
DAS |
Score à l'inclusionb |
5,5 |
5,7 |
5,5 |
|
Score semaine 52 b |
3,0 |
3,0 |
2,3 †, |
|
|
Rémissionc |
14 % |
18 % |
37 % †, |
|
|
HAQ |
Inclusion |
1,7 |
1,7 |
1,8 |
|
Semaine 52 |
1,1 |
1,0 |
0,8 †, |
a : les patients qui n'avaient pas terminé les 12 mois de
l'étude ont été considérés comme non-répondeurs.
b : les valeurs du Score
d'activité de la maladie (DAS) sont des moyennes.
c : la rémission est définie par un DAS < 1,6.
Valeurs de p lors des comparaisons appariées : † = p < 0,05 pour les comparaisons des groupes étanercept + méthotrexate vs méthotrexate et ϕ = p < 0,05 pour les comparaisons des groupes étanercept + méthotrexate vs étanercept.
L'évolution radiographique à 12 mois était significativement moins importante dans le groupe étanercept que dans le groupe méthotrexate, alors que l'association était significativement meilleure que chacune des monothérapies pour ralentir l'évolution radiographique (voir figure ci-dessous).
Évolution radiographique : comparaison d'étanercept vs méthotrexate vs étanercept associé au méthotrexate chez des patients ayant une PR d'ancienneté de 6 mois à 20 ans (résultats à 12 mois)
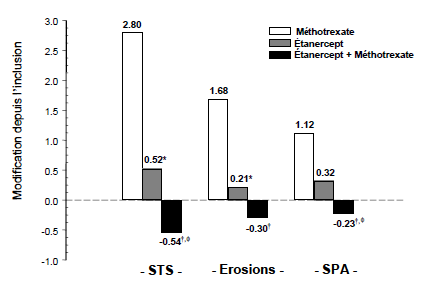
Valeurs de p lors des comparaisons appariées : * = p < 0,05 pour les comparaisons de l'étanercept vs méthotrexate, † = p < 0,05 pour les comparaisons de l'étanercept + méthotrexate vs méthotrexate et = p < 0,05 pour les comparaisons de l'étanercep + méthotrexate vs étanercept.
Des avantages significatifs avec l'étanercept associé au méthotrexate comparativement à l'étanercept en monothérapie et au méthotrexate en monothérapie ont également été observés après 24 mois. De même, des avantages significatifs avec l'étanercept en monothérapie comparativement au méthotrexate en monothérapie ont aussi été observés après 24 mois.
Dans une analyse où tous les patients sortis prématurément de l'étude quelle qu'en soit la raison étaient considérés comme s'étant aggravés, le pourcentage de patients sans aggravation (variation du STS ≤ 0,5) à 24 mois était plus élevé dans le groupe étanercept associé au méthotrexate, comparativement à l'étanercept en monothérapie et au méthotrexate en monothérapie (respectivement 62 %, 50 % et 36 % ; p < 0,05). La différence entre l'étanercept en monothérapie et le méthotrexate en monothérapie a également été significative (p < 0,05). Parmi les patients ayant terminé la totalité des 24 mois de traitement dans l'étude, les taux de patients sans aggravation étaient respectivement de 78 %, 70 % et 61 %.
La sécurité et l'efficacité de l'étanercept à la dose de 50 mg (deux injections de 25 mg par voie SC) administrée une fois par semaine ont été évaluées au cours d'une étude en double aveugle, contrôlée contre placebo chez 420 patients atteints de PR active. Dans cette étude, 53 patients ont reçu du placebo, 214 patients ont reçu 50 mg d'étanercept une fois par semaine et 153 patients ont reçu 25 mg d'étanercept deux fois par semaine. Les profils d'efficacité et de tolérance des deux schémas posologiques de l'étanercept ont été similaires à la 8ème semaine sur les signes et symptômes de la PR ; à la 16ème semaine, la comparabilité (non- infériorité) entre les deux schémas posologiques n'a pas été démontrée. Il a été démontré qu'une injection unique d'étanercept 50 mg/mL était bioéquivalente à deux injections simultanées de 25 mg/mL.
Patients adultes atteints derhumatisme psoriasique
L'efficacité de l'étanercept a été évaluée au cours d'une étude clinique randomisée, en double aveugle, contrôlée contre placebo, chez 205 patients atteints de rhumatisme psoriasique. Les patients étaient âgés de 18 à 70 ans et souffraient d'un rhumatisme psoriasique actif (≥ 3 articulations gonflées et ≥ 3 articulations douloureuses) dans au moins l'une de ces formes : (1) atteinte interphalangienne distale (AID) ; (2) polyarthrite (absence de nodules rhumatoïdes et présence de psoriasis) ; (3) arthropathie destructrice ; (4) rhumatisme psoriasique asymétrique ; ou (5) ankylose vertébrale de type inflammatoire. Les patients avaient également des plaques de psoriasis constituant une lésion dont le diamètre devait être ≥ 2 cm. Les patients étaient préalablement traités avec des AINS (86%), des DMARD (80 %), et des corticoïdes (24%). Les patients habituellement traités par méthotrexate (stable depuis ≥ 2 mois) pouvaient poursuivre le méthotrexate à une dose constante ≤ 25 mg/semaine. Des doses de 25 mg de l'étanercept (d'après les études de recherche de dose chez les patients atteints de polyarthrite rhumatoïde) ou de placebo étaient administrées par voie SC deux fois par semaine pendant 6 mois. A la fin de l'étude en double aveugle, les patients pouvaient entrer dans une étude d'extension en ouvert à long terme pour une durée totale allant jusqu'à 2 ans.
Les réponses cliniques ont été exprimées en pourcentages de patients atteignant une réponse ACR 20, 50 et 70 et en pourcentages d'amélioration du Critère de Réponse du Rhumatisme Psoriasique (PsARC). Les résultats sont résumés dans le tableau ci-après.
Réponses des patients atteints de rhumatisme psoriasique dans l'essai contrôlé contre placebo
|
|
|
Pourcentage de patients |
|
|
Réponse du rhumatisme psoriasique |
Placebo n = 104 |
Étanercepta n = 101 |
|
|
ACR 20 |
Mois 3 |
15 |
59b |
|
Mois 6 |
13 |
50b |
|
|
ACR 50 |
Mois 3 |
4 |
38b |
|
Mois 6 |
4 |
37b |
|
|
ACR 70 |
Mois 3 |
0 |
11b |
|
Mois 6 |
1 |
9c |
|
|
PsARC |
Mois 3 |
31 |
72b |
|
|
Mois 6 |
23 |
70b |
a : 25 mg étanercept SC deux fois par semaine b : p < 0,001, étanercept vs. placebo c : p < 0,01, étanercept vs. placebo
Parmi les patients atteints de rhumatisme psoriasique ayant reçu l'étanercept, les réponses cliniques étaient visibles dès la première visite (à 4 semaines) et se maintenaient pendant les 6 mois de traitement. L'étanercept a été significativement meilleur que le placebo sur tous les paramètres évaluant l'activité de la maladie (p < 0,001), et les réponses étaient similaires avec et sans traitement concomitant par le méthotrexate. La qualité de vie des patients atteints de rhumatisme psoriasique a été évaluée à plusieurs moments à l'aide de l'indice de handicap du questionnaire HAQ. L'indice de handicap était significativement amélioré à tous les temps d'évaluation chez les patients atteints de rhumatisme psoriasique traités par l'étanercept par rapport au groupe placebo (p < 0,001).
Les modifications radiographiques ont été évaluées dans l'étude sur le rhumatisme psoriasique. Des radiographies des mains et des poignets ont été réalisées à l'inclusion et aux mois 6, 12 et 24. Le STS modifié à 12 mois est présenté dans le tableau ci-dessous. Dans une analyse où tous les patients sortis de l'étude quelle qu'en soit la raison étaient considérés comme s'étant aggravés, le pourcentage de patients sans aggravation (variation du STS ≤ 0,5) à 12 mois était plus élevé dans le groupe étanercept comparativement au groupe placebo (respectivement 73 % vs 47 %, p ≤ 0,001). L'effet de l'étanercept sur l'aggravation radiographique était maintenu chez les patients qui poursuivaient le traitement au cours de la deuxième année. Le ralentissement des dommages structuraux articulaires périphériques était observé chez des patients ayant une atteinte polyarticulaire symétrique.
Évolution annualisée moyenne (ET) du score total de sharp depuis l'inclusion
|
|
Placebo |
Étanercept |
|
Temps |
(n = 104) |
(n = 101) |
|
Mois 12 |
1,00 (0,29) |
- 0,03 (0,09)a |
ET = erreur type. a. p = 0,0001.
Les capacités fonctionnelles ont été améliorées avec le traitement par l'étanercept pendant la période en double aveugle, et ce bénéfice a été maintenu au cours de l'exposition à long terme jusqu'à 2 ans.
Dans la forme axiale du rhumatisme psoriasique, proche de la spondylarthrite ankylosante, et dans la forme mutilante, les preuves d'efficacité de l'étanercept sont insuffisantes en raison du nombre trop faible de patients étudiés.
Aucune étude n'a été effectuée chez des patients atteints de rhumatisme psoriasique avec le schéma posologique de 50 mg une fois par semaine. Les preuves de l'efficacité du schéma posologique d'une fois par semaine dans cette population de patients reposent sur des données provenant d'une étude chez des patients atteints de spondylarthrite ankylosante.
Patients adultes atteints de spondylarthrite ankylosante
L'efficacité de l'étanercept dans la spondylarthrite ankylosante a été évaluée dans 3 études, randomisées, en double aveugle, ayant comparé l'administration à deux fois par semaine d'étanercept 25 mg versus placebo. Au total, 401 patients ont été recrutés dont 203 étaient traités par l'étanercept. La plus importante de ces études (n = 277) a recruté des patients âgés de 18 à 70 ans et qui avaient une spondylarthrite ankylosante active définie par des scores d'échelle visuelle analogique (EVA) ≥ 30 pour la durée et l'intensité moyennes de la raideur matinale, associée à des scores EVA ≥ 30 pour au moins 2 des 3 paramètres suivants : évaluation globale par le patient ; moyenne des valeurs EVA pour la dorsalgie nocturne et la dorsalgie totale ; moyenne des 10 questions de l'Indice Fonctionnel de la Spondylarthrite Ankylosante de Bath (BASFI). Les patients recevant des DMARD, des AINS ou des corticoïdes pouvaient continuer ces traitements à des doses stables. Les patients présentant une ankylose complète de la colonne vertébrale n'ont pas été inclus dans l'étude. Des doses de 25 mg d'étanercept (déterminées lors des études de recherche de dose chez les patients atteints de polyarthrite rhumatoïde) ou de placebo ont été administrées par voie sous-cutanée deux fois par semaine pendant 6 mois chez 138 patients.
Le critère principal d'efficacité (ASAS 20) consistait en une amélioration ≥ 20 % d'au moins 3 des 4 domaines du critère ASAS (Assessment in Ankylosing Spondylitis : évaluations globales par le patient, dorsalgie, BASFI, et inflammation) et en l'absence d'aggravation du domaine restant. Les réponses ASAS 50 et ASAS 70 consistaient en des améliorations respectives de 50 % et 70 % sur les mêmes critères.
Par rapport au placebo, le traitement par l'étanercept a démontré des améliorations significatives des réponses ASAS 20, ASAS 50 et ASAS 70 dès la deuxième semaine après l'initiation du traitement.
Réponses des patients atteints de spondylarthrite ankylosante au cours d'un essai contrôlé contre placebo
|
|
Pourcentage de Patients |
|
|
Réponse de la Spondylarthrite Ankylosante |
Placebo n = 139 |
Étanercept n = 138 |
|
ASAS 20 |
|
|
|
2 semaines |
22 |
46a |
|
3 mois |
27 |
60a |
|
6 mois |
23 |
58a |
|
ASAS 50 |
|
|
|
2 semaines |
7 |
24a |
|
3 mois |
13 |
45a |
|
6 mois |
10 |
42a |
|
ASAS 70 |
|
|
|
2 semaines |
2 |
12b |
|
3 mois |
7 |
29b |
|
6 mois |
5 |
28b |
a : p < 0,001, étanercept vs placebo
b : p = 0,002, étanercept vs placebo
Parmi les patients atteints de spondylarthrite ankylosante ayant reçu l'étanercept, les réponses cliniques sont apparues dès la première visite (2 semaines) et se sont maintenues au cours des 6 mois de traitement. Les réponses étaient similaires chez les patients qui, à l'inclusion, recevaient ou non des traitements concomitants.
Des résultats similaires ont été obtenus au cours des 2 essais d'effectifs moins importants réalisés dans la spondylarthrite ankylosante.
Dans une quatrième étude, la sécurité et l'efficacité de l'étanercept 50 mg (deux injections SC de 25 mg) administré une fois par semaine versus étanercept 25 mg administré deux fois par semaine ont été évaluées au cours d'une étude en double aveugle, contrôlée contre placebo menée chez 356 patients atteints de spondylarthrite ankylosante active. Les profils de sécurité et d'efficacité des schémas posologiques de 50 mg une fois par semaine et de 25 mg deux fois par semaine ont été similaires. Patients adultes atteints de spondyloarthrite axiale non radiographique
Étude 1
L'efficacité de l'étanercept dans la spondyloarthrite axiale non radiographique (SpA axiale NR) a été évaluée au cours d'une étude randomisée, contrôlée contre placebo, d'une durée de 12 semaines en double aveugle. L'étude a évalué 215 patients adultes (population en intention de traiter modifiée) atteints de SpA axiale NR active (âge : 18 à 49 ans), définis comme répondant aux critères de classification ASAS pour la spondyloarthrite axiale mais ne répondant pas aux critères New York modifiés pour la SA. Les patients devaient également présenter une réponse inadéquate ou une intolérance à deux AINS ou plus. Au cours de la période de traitement en double aveugle, les patients ont reçu 50 mg d'étanercept par semaine ou un placebo pendant 12 semaines. Le critère principal d'efficacité (ASAS 40) consistait en une amélioration de 40 % d'au moins trois des quatre domaines ASAS sans aggravation du domaine restant. La période de traitement en double aveugle était suivie d'une période de traitement en ouvert au cours de laquelle tous les patients ont reçu 50 mg d'étanercept par semaine pendant une durée complémentaire pouvant aller jusqu'à 92 semaines. Des IRM de l'articulation sacro-iliaque et de la colonne vertébrale ont été réalisées pour évaluer l'inflammation à l'inclusion et aux semaines 12 et 104.
Par rapport au placebo, le traitement par l'étanercept a montré une amélioration statistiquement significative des réponses ASAS 40, ASAS 20 et ASAS 5/6. Une amélioration significative de la rémission partielle ASAS et de la réponse BASDAI 50 a également été observée. Les résultats obtenus à la semaine 12 sont présentés dans le tableau ci-dessous.
Réponse d'efficacité dans l'étude sur la SpA axiale NR contrôlée contre placebo : pourcentage de patients ayant atteint les critères d'évaluation
*Des données complètes n'ont pas pu être obtenues pour chaque critère d'évaluation chez tous les patients
**ASAS = Assessments in Spondyloarthritis
International Society
*** Bath Ankylosing Spondylitis Disease Activity Index
a : p < 0,001, b : < 0,01 et c : < 0,05 respectivement entre l'étanercept et le placebo
A la semaine 12, une amélioration statistiquement significative du score SPARCC (Spondyloarthritis Research Consortium of Canada) pour l'articulation sacro-iliaque (ASI) mesurée par IRM a été observée chez les patients recevant l'étanercept. La variation moyenne ajustée par rapport à la valeur initiale était de 3,8 pour les patients traités par l'étanercept (n = 95) contre 0,8 pour les patients ayant reçu le placebo (n = 105) (p < 0,001). A la semaine 104, la variation moyenne par rapport à la valeur initiale du score SPARCC mesuré par IRM pour tous les patients traités par l'étanercept était de 4,64 pour l'ASI (n = 153) et de 1,40 pour la colonne vertébrale (n = 154).
Par rapport au placebo, l'étanercept a permis une amélioration significativement plus importante de la plupart des évaluations de la capacité fonctionnelle et de la qualité de vie liées à la santé entre l'inclusion et la semaine 12, y compris le BASFI (Bath Ankylosing Spondylitis Functional Index), le score sur l'état de santé général EuroQol 5D et la composante physique du score SF-36.
Chez les patients atteints de SpA axiale NR traités par l'étanercept, les réponses cliniques étaient visibles au moment de la première visite (2 semaines) et ont été maintenues pendant les 2 ans de traitement. Les améliorations de la qualité de vie et de la fonction physique ont été également maintenues pendant les 2 ans de traitement. Les données de ces 2 années n'ont pas révélé de nouveaux signaux de sécurité. À la semaine 104, 8 sujets ont progressé vers un score bilatéral de grade 2 par radiographie de la colonne vertébrale selon les critères radiographiques de New York modifiés, indicateurs de spondyloarthropathie axiale.
Étude 2
Cette étude sur 3 périodes, multicentrique, en ouvert, de phase IV, a permis d'évaluer l'arrêt et la reprise du traitement par etanercept chez des patients atteints de SpA axiale NR active ayant obtenu une réponse adéquate (maladie inactive définie par un score d'activité de la maladie de la spondylarthrite ankylosante [ASDAS — Ankylosing Spondylitis Disease Activity Score] protéine C réactive [CRP] inférieur à 1,3) après 24 semaines de traitement.
209 patients adultes atteints de SpA axiale NR active (âge : 18 à 49 ans), définis comme des patients répondant aux critères de classification de la Société internationale de spondylarthrite (ASAS) de la spondyloarthrite axiale (mais ne répondant pas aux critères de New York modifiés pour la SA), présentant des résultats positifs à l'IRM (inflammation active à l'IRM suggérant fortement une sacroiliite associée à une SpA) et/ou une CRP-hs positive (définie comme une protéine C réactive ultrasensible [CRP-hs] > 3 mg/l) et des symptômes actifs définis par un ASDAS-CRP supérieur ou égal à 2,1 lors de la visite de sélection, ont reçu 50 mg d' etanercept par semaine en ouvert ainsi qu'un traitement de fond par AINS à dose stable, à dose anti-inflammatoire optimale tolérée pendant 24 semaines durant la Période 1. Les patients devaient également présenter une réponse inadéquate ou une intolérance à deux AINS ou plus. À la semaine 24, 119 (57 %) patients ont atteint le stade de maladie inactive et sont entrés dans la phase d'arrêt de traitement de 40 semaines de la Période 2, au cours de laquelle les sujets ont arrêté le traitement par etanercept, tout en conservant l'AINS en traitement de fond. La principale mesure de l'efficacité était la survenue d'une poussée (définie par un ASDAS-vitesse de sédimentation (VS) supérieur ou égal à 2,1) dans les 40 semaines suivant l'arrêt d'etanercept. Les patients qui ont présenté des poussées ont été retraités avec etanercept à raison de 50 mg par semaine pendant 12 semaines (Période 3).
Au cours de la Période 2, la proportion de patients ayant présenté ≥ 1 poussée est passée de 22 % (25/112) à la semaine 4 à 67 % (77/115) à la semaine 40. Dans l'ensemble, 75 % (86/115) des patients ont présenté une poussée à différents moments au cours des 40 semaines suivant l'arrêt d'etanercept.
L'objectif secondaire principal de l'Étude 2 était le délai d'apparition d'une poussée après l'arrêt d'etanercept ainsi que le délai d'apparition d'une poussée chez les patients de l'Étude 1 ayant satisfait aux critères d'éligibilité de la phase d'arrêt de l'Étude 2 et ayant poursuivi le traitement par etanercept.
Le délai médian d'apparition d'une poussée après l'arrêt d'etanercept était de 16 semaines (IC à 95 % : 13-24 semaines). Moins de 25 % des patients de l'Étude 1 qui n'ont pas eu d'arrêt de traitement ont présenté une poussée sur l'équivalent de 40 semaines comme dans la Période 2 de l'Étude 2. Le délai d'apparition d'une poussée était significativement plus court chez les sujets ayant arrêté le traitement par etanercept (Étude 2) que chez ceux ayant reçu un traitement continu par etanercept (Étude 1), p < 0,0001.
Sur les 87 patients entrés dans la Période 3 et ayant été retraités par etanercept à raison de 50 mg par semaine pendant 12 semaines, 62 % (54/87) ont de nouveau atteint le stade de maladie inactive, et 50 % d'entre eux l'ont atteint en 5 semaines (IC à 95 % : 4-8 semaines).
Patients adultes atteints de psoriasis en plaques
L'utilisation de l'étanercept est recommandée chez les patients définis à la rubrique Indications thérapeutiques. Les patients « en échec » dans la population cible sont définis comme présentant une réponse insuffisante (PASI < 50 ou PGA insatisfaisant), ou une aggravation de la maladie au cours du traitement avec au moins l'un des trois traitements systémiques majeurs disponibles utilisés à une posologie adéquate pendant une durée suffisamment longue pour évaluer la réponse au traitement.
L'efficacité de l'étanercept versus les autres traitements systémiques chez les patients avec un psoriasis modéré à sévère (répondeurs aux autres traitements systémiques) n'a pas été évaluée dans des études comparant directement l'étanercept aux autres traitements systémiques. A la place, la sécurité et l'efficacité de l'étanercept ont été évaluées au cours de quatre études randomisées, en double aveugle, contrôlées contre placebo. Le critère d'évaluation principal de l'efficacité dans les quatre études était la proportion de patients dans chaque groupe de traitement qui atteignait le PASI 75 (c'est-à-dire une amélioration par rapport à l'inclusion d'au moins 75% du score Psoriasis Area and Severity Index) à 12 semaines.
L'Étude 1 était une étude de phase II chez des patients âgés d'au moins 18 ans et présentant un psoriasis en plaques actif mais cliniquement stable atteignant au moins 10 % de la surface corporelle. Cent douze (112) patients ont été randomisés pour recevoir une dose de 25 mg d'étanercept (n = 57) ou du placebo (n = 55) deux fois par semaine pendant 24 semaines.
L'Étude 2 a évalué 652 patients atteints de psoriasis en plaques chronique avec les mêmes critères d' inclusion que dans l'étude 1 et un score PASI ≥ 10 au moment de la sélection. L'étanercept a été administré à des doses de 25 mg une fois par semaine, 25 mg deux fois par semaine ou 50 mg deux fois par semaine pendant 6 mois consécutifs. Au cours des 12 premières semaines de la période de traitement en double aveugle, les patients ont reçu du placebo ou l'une des trois doses d'étanercept décrites ci-dessus. Après 12 semaines de traitement, les patients du groupe placebo ont commencé le traitement en aveugle par l'étanercept (25 mg deux fois par semaine) ; les patients dans les groupes de traitement actif ont continué jusqu'à la semaine 24, à la dose à laquelle ils avaient été initialement randomisés.
L'Étude 3 a évalué 583 patients et les critères d'inclusion étaient les mêmes que dans l'Étude 2. Dans cette étude, les patients ont reçu une dose de 25 mg ou de 50 mg d'étanercept, ou un placebo, deux fois par semaine pendant 12 semaines ; puis tous les patients ont reçu 25 mg d'étanercept deux fois par semaine en ouvert pendant 24 semaines supplémentaires.
L'Étude 4 a évalué 142 patients et les critères d'inclusion étaient similaires à ceux des Études 2 et 3. Dans cette étude, les patients ont reçu une dose de 50 mg d'étanercept ou un placebo une fois par semaine pendant 12 semaines ; puis tous les patients ont reçu 50 mg d'étanercept une fois par semaine en ouvert pendant 12 semaines supplémentaires.
Dans l'Étude 1, le groupe traité par l'étanercept avait une proportion significativement plus élevée de patients présentant une réponse PASI 75 à la semaine 12 (30 %) par rapport au groupe sous placebo (2 %) (p < 0,0001). A 24 semaines, 56 % des patients dans le groupe traité par l'étanercept avaient atteint le PASI 75 par rapport à 5 % des patients sous placebo. Les résultats principaux des Études 2, 3 et 4 sont présentés ci-dessous.
Réponses des patients atteints de psoriasis dans les études 2, 3 et 4
|
|
------------------ Étude 2--------------- |
--------------- Étude 3----------- |
-------------- Étude 4--------- |
|||||||
|
Placebo |
------ 25 mg 2 fois/sem |
Étanercept------ 50 mg 2 fois/sem |
Placebo |
----Étanercept 25 mg 2 fois/ sem |
---- 50 mg 2 fois/ sem |
Placebo |
---Étanercept 50 mg 1 fois/ sem |
---- 50 mg 1 fois/ Sem |
||
|
|
n = 166 |
n = n = 162 162 |
n = 164 |
n = 164 |
n = 193 |
n = 196 |
n = 196 |
n = 46 |
n = 96 |
n = 90 |
|
Réponse (%) |
sem 12 |
sem sem 12 24a |
sem 12 |
sem 24a |
sem 12 |
sem 12 |
sem 12 |
sem 12 |
sem 12 |
sem 24a |
|
PASI 50 |
14 |
58* 70 |
74* |
77 |
9 |
64* |
77* |
9 |
69* |
83 |
|
PASI 75 |
4 |
34* 44 |
49* |
59 |
3 |
34* |
49* |
2 |
38* |
71 |
|
DSGAb, pas de lésions apparentes ou presque pas de lésions apparentes |
5 |
34* 39 |
49* |
55 |
4 |
39* |
57* |
4 |
39* |
64 |
* p ≤ 0,0001 comparé au placeboa : Aucune comparaison statistique versus placebo n'a été faite à la semaine 24 dans les Études 2 et 4 étant donné que le groupe initialement sous placebo a commencé à recevoir l'étanercept 25 mg deux fois/sem ou 50 mg une fois/sem à partir de la semaine 13 jusqu'à la semaine 24.
b : Dermatologist Static Global Assessment. Pas de lésions apparentes ou presque pas de lésions apparentes, défini par 0 ou 1 sur une échelle de 0 à 5.
Parmi les patients atteints de psoriasis en plaques qui recevaient l'étanercept, des réponses significatives comparativement au placebo sont apparues à la première visite (2 semaines) et ont été maintenues durant les 24 semaines de traitement.
L'Étude 2 comprenait également une période d'arrêt du traitement au cours de laquelle les patients qui avaient atteint une amélioration du PASI d'au moins 50 % à la semaine 24 arrêtaient le traitement. L'apparition d'un rebond (PASI ≥ 150 % de la valeur à l'inclusion) et le délai de rechute (définie par la perte d'au moins la moitié de l'amélioration obtenue entre l'inclusion et la semaine 24) ont été évalués chez les patients qui n'étaient plus sous traitement. Au cours de la période sans traitement, les symptômes du psoriasis sont progressivement réapparus avec un délai médian de rechute de 3 mois. Aucun effet rebond de la maladie et aucun événement indésirable grave lié au psoriasis n'ont été observés. Il existe des données démontrant le bénéfice de la reprise du traitement par l'étanercept chez les patients qui répondaient initialement au traitement.
Dans l'Étude 3, la majorité des patients (77 %) qui étaient initialement randomisés à la dose de 50 mg deux fois par semaine et avaient vu leur dose d'étanercept abaissée à 25 mg deux fois par semaine à la semaine 12 ont eu une réponse PASI 75 maintenue jusqu'à la semaine 36. Pour les patients qui recevaient 25 mg deux fois par semaine tout au long de l'étude, la réponse PASI 75 continuait de s'améliorer entre les semaines 12 et 36.
Dans l'Étude 4, le groupe traité par l'étanercept avait une proportion plus élevée de patients avec une réponse PASI 75 à la semaine 12 (38 %) par rapport au groupe recevant le placebo (2 %) (p < 0,0001). Pour les patients qui recevaient 50 mg une fois par semaine tout au long de l'étude, les réponses d'efficacité ont continué à s'améliorer avec un PASI 75 à la semaine 24 atteignant 71 %.
Dans les études à long terme (jusqu'à 34 mois) et en ouvert au cours desquelles l'étanercept a été administré sans interruption, les réponses cliniques étaient maintenues et la sécurité était comparable aux études à court terme.
Une analyse des données des essais cliniques n'a révélé aucune caractéristique de la maladie à l'inclusion qui pourrait conduire les cliniciens à sélectionner le type de posologie le plus approprié (intermittent ou continu). Par conséquent, le choix d'un traitement intermittent ou continu doit reposer sur le jugement du médecin et les besoins individuels des patients.
Anticorps anti-étanercept
Des anticorps anti-etanercept ont été détectés dans le sérum de certains sujets traités par etanercept. Ces anticorps ont tous été non-neutralisants et sont généralement transitoires. Il semble qu'il n'y a aucune corrélation entre le développement d'anticorps et une réponse clinique ou des évènements indésirables.
Population pédiatrique
Patients pédiatriques atteints d'arthrite juvénile idiopathique
La sécurité et l'efficacité de l'étanercept ont été évaluées au cours d'une étude en deux parties, de 69 enfants présentant une arthrite juvénile idiopathique d'évolution polyarticulaire avec différentes formes de début de la maladie (polyarthrite, pauciarthrite, origine systémique). Les patients inclus dans l'étude étaient âgés de 4 à 17 ans avec une arthrite juvénile idiopathique d'évolution polyarticulaire d'intensité modérée à sévère, réfractaires ou intolérants au méthotrexate. Une dose stable d'un seul anti-inflammatoire non stéroïdien et/ou de prednisone (< 0,2 mg/kg/jour ou 10 mg maximum) a été maintenue chez les patients. Dans la première partie de l'étude, tous les patients ont reçu 0,4 mg/kg (maximum 25 mg par injection) d'étanercept administré par voie sous-cutanée deux fois par semaine. Dans la deuxième partie de l'étude, les patients avec une réponse clinique à 90 jours ont été randomisés pour soit rester sous étanercept, soit recevoir un placebo pendant quatre mois avec évaluation de la rechute clinique. Les réponses ont été mesurées en utilisant l'ACR Pedi 30, définie par une amélioration ≥ 30 % d'au moins trois des six critères de base du score ACR Pedi et une aggravation ≥ 30 % d'au plus un de ces critères, incluant le nombre d'articulations atteintes, la limitation des mouvements, les évaluations globales par le médecin et par le patient/parent, le handicap fonctionnel et la vitesse de sédimentation (vs). La rechute clinique était définie comme une aggravation ≥ 30 % de trois des six critères de base du score ACR Pedi et une amélioration ≥ 30 % d'au plus un de ces critères ainsi qu'un minimum de deux articulations atteintes.
Dans la première partie de l'étude, 51 des 69 patients (74 %) ont bénéficié d'une réponse clinique et ont été inclus dans la deuxième partie de l'étude. Dans la deuxième partie de l'étude, 6 des 25 patients (24%) maintenus sous étanercept ont eu une rechute clinique en comparaison à 20 sur 26 patients (77%) sous placebo (p = 0,007). A partir du début de la deuxième partie de l'étude, la médiane du délai de rechute clinique a été supérieure ou égale à 116 jours pour les patients ayant reçu l'étanercept et 28 jours pour les patients sous placebo. Parmi les patients ayant bénéficié d'une réponse clinique à 90 jours et ayant été inclus dans la deuxième partie de l'étude, certains des patients maintenus sous étanercept ont continué à s'améliorer entre le troisième mois et le septième mois alors que ceux sous placebo ne se sont pas améliorés.
Dans une étude d'extension de sécurité, en ouvert, 58 patients pédiatriques de l'étude mentionnée cidessus (à partir de l'âge de 4 ans au moment du recrutement) ont continué à recevoir l'étanercept pour une durée allant jusqu'à 10 ans. Les taux d'événements indésirables graves et d'infections graves n'ont pas augmenté avec une exposition à long terme.
La sécurité à long terme d'une monothérapie par l'étanercept (n = 103), de l'étanercept plus méthotrexate (n = 294) ou d'une monothérapie par le méthotrexate (n = 197) a été évaluée pendant une période maximale de 3 ans dans un registre de 594 enfants âgés de 2 à 18 ans présentant une arthrite juvénile idiopathique, parmi lesquels 39 étaient âgés de 2 à 3 ans. En général, les infections ont été plus fréquemment rapportées chez les patients traités par étanercept comparé au méthotrexate seul (3,8 contre 2 %), et les infections associées à l'utilisation d'étanercept ont été de nature plus sévère.
Dans une autre étude en ouvert à bras unique (n = 127), 60 patients atteints d'une oligoarthrite extensive (OE) (15 patients âgés de 2 à 4 ans, 23 patients âgés de 5 à 11 ans et 22 patients âgés de 12 à 17 ans), 38 patients atteints d'une arthrite liée à l'enthésite (âgés de 12 à 17 ans), et 29 patients atteints d'une arthrite psoriasique (âgés de 12 à 17 ans) ont été traités par l'étanercept à une dose de 0,8 mg/kg (50 mg maximum par injection) administrée une fois par semaine pendant 12 semaines. Dans chaque sous-type d'AJI, la majorité des patients a répondu aux critères de l'ACR Pedi 30 et a montré une amélioration clinique des critères d'évaluation secondaires tels que le nombre d'articulations douloureuses et l'évaluation globale du médecin. Le profil de sécurité est similaire à celui observé dans les autres études dans l'AJI.
Sur les 127 patients de l'étude mère, 109 ont participé à l'étude d'extension en ouvert et ont fait l'objet d'un suivi pendant 8 années supplémentaires, soit une durée totale allant jusqu'à 10 ans. À la fin de l'étude d'extension, 84/109 (77 %) patients avaient terminé l'étude ; 27 (25 %) prenaient activement de l'étanercept, 7 (6 %) avaient arrêté le traitement en raison d'une maladie faible/inactive ; 5 (5 %) avaient repris l'étanercept après un arrêt antérieur du traitement ; et 45 (41 %) avaient arrêté l'étanercept (mais restaient en observation) ; 25/109 (23 %) patients ont définitivement quitté l'étude. Les améliorations de l'état clinique obtenues dans l'étude mère ont été généralement maintenues pour tous les critères d'efficacité pendant toute la période de suivi. Les patients prenant activement de l'étanercept ont pu entrer dans une période d'arrêt-retraitement facultative une fois au cours de l'étude d'extension, en fonction de la réponse clinique évaluée par l'investigateur. 30 patients sont entrés dans la période d'arrêt du traitement. Il a été rapporté que 17 patients avaient présenté une poussée (définie comme une aggravation ≥ 30 % d'au moins 3 des 6 critères de l'ACR Pedi avec une amélioration ≥ 30 % d'au plus 1 des 6 critères restants et un minimum de 2 articulations atteintes) ; le délai médian de poussée après l'arrêt de l'étanercept a été de 190 jours. 13 patients ont été re-traités et le délai médian de re-traitement après l'arrêt a été estimé à 274 jours. En raison du faible nombre de patients, ces résultats doivent être interprétés avec prudence.
Le profil de sécurité est similaire à celui observé dans l'étude mère.
Aucune étude n'a été réalisée chez les patients atteints d'une arthrite juvénile idiopathique pour évaluer les effets de la poursuite du traitement par l'étanercept chez les patients non répondeurs dans un délai de 3 mois après l'initiation du traitement par l'étanercept. En outre, aucune étude n'a été réalisée pour évaluer les effets de la diminution de la dose recommandée d'étanercept après une utilisation à long terme chez les patients atteints d'une AJI.
Patients pédiatriques atteints de psoriasis en plaques
L'efficacité de l'étanercept a été évaluée au cours d'une étude randomisée, en double aveugle, contrôlée contre placebo réalisée chez 211 patients pédiatriques âgés de 4 à 17 ans atteints de psoriasis en plaques modéré à sévère (défini par un score sPGA ≥ 3, une surface cutanée atteinte ≥ 10%, et un PASI ≥ 12). Les patients éligibles avaient déjà reçu un traitement par photothérapie ou un traitement systémique, ou étaient mal contrôlés par un traitement topique.
Les patients ont reçu soit l'étanercept 0,8 mg/kg (jusqu'à 50 mg) soit le placebo une fois par semaine pendant 12 semaines. A la semaine 12, davantage de patients étaient répondeurs (exemple : PASI 75) dans le groupe étanercept comparativement au groupe placebo.
Résultats à 12 semaines dans le psoriasis en plaques pediatrique
|
|
Étanercept 0.8 mg/kg une fois par semaine (n = 106) |
Placebo (n = 105) |
|
PASI 75, n (%) |
60 (57 %)a |
12 (11 %) |
|
PASI 50, n (%) |
79 (75 %)a |
24 (23 %) |
|
sPGA « blanchi »” ou « quasi blanchi », n (%) |
56 (53 %)a |
14 (13 %) |
Abréviation: sPGA-évaluation globale statique par le médecin a : p < 0,0001 comparé au placebo.
Après la période de traitement en double aveugle de 12 semaines, tous les patients ont reçu l'étanercept 0,8 mg/kg (jusqu'à 50 mg) une fois par semaine pendant 24 semaines supplémentaires. Les réponses observées pendant la période en ouvert étaient similaires à celles observées pendant la période en double aveugle.
Pendant une période randomisée d'arrêt, un nombre significativement plus élevé de patients rerandomisés dans le groupe placebo a présenté une rechute (perte de la réponse au PASI 75) en comparaison aux patients re-randomisés dans le groupe étanercept. En traitement continu, les réponses étaient maintenues jusqu'à 48 semaines.
La sécurité et l'efficacité à long terme de l'étanercept 0,8 mg/kg (jusqu'à 50 mg) une fois par semaine ont été évaluées dans une étude d'extension en ouvert chez 181 sujets pédiatriques atteints de psoriasis en plaques pendant une période maximale de 2 ans après l'étude de 48 semaines décrite ci-dessus. L'expérience à long terme avec l'étanercept était généralement comparable à celle de l'étude initiale de 48 semaines et n'a révélé aucune nouvelle donnée de sécurité.
Les concentrations sériques d'etanercept ont été déterminées par la méthode immunoenzymatique(Enzyme-Linked Immunosorbent Assay, ELISA), qui détecte les produits de dégradation réagissant au dosage ELISA ainsi que la molécule mère.
Absorption
L'étanercept est absorbé lentement depuis le site d'injection sous-cutanée, atteignant une concentration maximale environ 48 heures après administration unique. La biodisponibilité absolue est de 76 %. Avec deux doses par semaine, on peut s'attendre à ce que les concentrations à l'état d'équilibre représentent environ deux fois celles que l'on mesure après administration unique. Après l'administration unique, par voie sous-cutanée, de 25 mg d'étanercept, la concentration sérique maximale moyenne observée chez les volontaires sains était de 1,65 ± 0,66 µg/mL, l'aire sous la courbe étant de 235 ± 96,6 µg.h/mL.
Les caractéristiques des concentrations sériques moyennes à l'état d'équilibre chez les patients atteints de PR étaient une Cmax de 2,4 mg/l vs 2,6 mg/l, une Cmin de 1,2 mg/l vs 1,4 mg/l, et une aire sous la courbe partielle (ASC) de 297 mgh/l vs 316 mgh/l pour la dose de 50 mg d'étanercept une fois par semaine (n = 21) vs 25 mg d'étanercept deux fois par semaine (n = 16) respectivement. Dans une étude en ouvert, croisée, avec administration unique de deux posologies différentes chez des volontaires sains, il a été démontré que l'administration d'une injection unique de 50 mg/mL d'étanercept était bioéquivalente à deux injections simultanées de 25 mg/mL.
Dans une analyse pharmacocinétique de patients atteints de spondylarthrite ankylosante, les ASC de l'étanercept à l'équilibre ont été de 466 gh/mL et de 474 gh/mL respectivement pour l'étanercept50 mg une fois par semaine (n = 154) et pour l'étanercept 25 mg deux fois par semaine (n = 148).
Distribution
La courbe de concentration d'étanercept en fonction du temps est biexponentielle. Le volume de distribution central d'étanercept est de 7,6 l, alors que le volume de distribution à l'état d'équilibre est de 10,4 l.
Élimination
L'étanercept est éliminé lentement par l'organisme. Sa demi-vie est longue, environ 70 heures. Sa clairance est d'environ 0,066 l/h chez les patients atteints de polyarthrite rhumatoïde, soit un peu moins que celle observée chez les volontaires sains (0,11 l/h). De plus, la pharmacocinétique de l'étanercept chez les patients atteints de polyarthrite rhumatoïde, de spondylarthrite ankylosante ou de psoriasis en plaques est similaire.
Il n'existe apparemment aucune différence pharmacocinétique entre les hommes et les femmes.
Linéarité
La recherche d'une proportionnalité par rapport à la dose administrée n'a pas fait l'objet d'une évaluation particulière, mais il n'existe aucun signe de saturation de la clairance dans l'intervalle des posologies proposées.
Populations particulières
Insuffisance rénale
Bien que l'on détecte de la radioactivité dans les urines après l'administration d'étanercept radiomarqué à des patients et à des volontaires sains, aucune augmentation des concentrations d'étanercept n'a été observée chez les patients présentant une insuffisance rénale aiguë. La présence d'une insuffisance rénale ne devrait pas nécessiter d'ajustement de la posologie.
Insuffisance hépatique
Aucune augmentation des concentrations d'étanercept n'a été observée chez les patients présentant une insuffisance hépatique aiguë. La présence d'une insuffisance hépatique ne devrait pas nécessiter d'ajustement de la posologie.
Personnes âgées
L'analyse des concentrations sériques d'etanercept dans le cadre des études de pharmacocinétique de population a été étudiée pour mesurer l'impact du grand âge. La clairance et le volume estimés chez les patients âgés de 65 à 87 ans étaient comparables aux estimations obtenues chez les patients âgés de moins de 65 ans.
Population pédiatrique
Patients pédiatriques atteints d'arthrite juvénile idiopathique
Dans un essai de l'étanercept dans l'arthrite juvénile idiopathique d'évolution polyarticulaire, 69 patients (âgés de 4 à 17 ans) ont reçu 0,4 mg d'étanercept/kg deux fois par semaine pendant trois mois. Les courbes de concentration sériques étaient similaires à celles que l'on observe chez les patients adultes atteints de polyarthrite rhumatoïde. Les enfants les plus jeunes (âgés de 4 ans) ont eu une clairance réduite (clairance augmentée quand elle est ajustée au poids) par rapport aux enfants plus âgés (âgés de 12 ans) et aux adultes. Une modélisation des dosages suggère que les enfants plus âgés (10 à 17 ans) auront des taux sériques proches de ceux des adultes, et que les plus jeunes enfants auront des taux notablement plus bas.
Patients pédiatriques atteints de psoriasis en plaques
Les patients atteints de psoriasis en plaques pédiatrique (âgés de 4 à 17 ans) ont reçu 0,8 mg/kg (jusqu'à une dose maximale de 50 mg par semaine) d'etanercept une fois par semaine jusqu'à 48 semaines. Les concentrations sériques moyennes à l'état d'équilibre ont varié de 1,6 à 2,1 µg/ml aux semaines 12, 24 et 48. Ces concentrations moyennes chez les patients atteints de psoriasis en plaques pédiatriques ont été similaires aux concentrations observées chez les patients atteints d'arthrite juvénile idiopathique (traités par 0,4 mg/kg d'étanercept deux fois par semaine, jusqu'à une dose maximale de 50 mg par semaine). Ces concentrations moyennes étaient similaires à celles observées chez les patients adultes atteints de psoriasis en plaques traités par 25 mg d'étanercept deux fois par semaine.
Erelzi n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Les études de toxicité de l'étanercept n'ont fait apparaître aucune dose limite toxique ou de toxicité vis-à-vis d'un organe cible. Au cours d'une batterie de tests réalisés in vitro et in vivo, l'étanercept a été considéré comme non génotoxique. Les études de carcinogénicité, et les évaluations standard de fertilité et de toxicité postnatales, n'ont pas pu être réalisées avec l'étanercept à cause du développement d'anticorps neutralisants chez les rongeurs.
L'étanercept n'a pas induit de mortalité ou de signe notable de toxicité chez la souris ou le rat à la dose de 2000 mg/kg en administration unique par voie sous-cutanée ou à la dose de 1000 mg/kg en injection unique par voie intraveineuse. Aucune dose limite toxique de l'étanercept ou de toxicité visà-vis d'un organe cible chez le singe cynomolgus n'ont été mises en évidence après administration deux fois par semaine, par voie sous-cutanée, pendant 4 à 26 semaines consécutives d'une dose de 15 mg/kg. Cette dose correspond à une ASC des concentrations plasmatiques 27 fois supérieure à celle obtenue chez l'homme à la dose recommandée de 25 mg.
Instructions pour l'utilisation et la manipulation de la seringue préremplie d'Erelzi
Avant l'injection, la seringue préremplie à usage unique d'Erelzi doit atteindre la température ambiante (environ 15 à 30 minutes). Le capuchon d'aiguille ne doit pas être retiré pendant que la seringue atteint la température ambiante. La solution doit être limpide à légèrement opalescente, incolore à jaune pâle, et peut contenir des petites particules de protéines translucides ou blanches.
Des instructions complètes pour l'administration sont données dans la notice, à la rubrique 7 « Instructions pour l'utilisation de la seringue préremplie d'Erelzi ».
Instructions pour l'utilisation et la manipulation du stylo prérempli d'Erelzi
Avant l'injection, les stylos préremplis à usage unique d'Erelzi doivent atteindre la température ambiante (environ 15 à 30 minutes). Le capuchon d'aiguille ne doit pas être retiré pendant que le stylo atteint la température ambiante. En regardant par la fenêtre de visualisation, la solution doit être limpide à légèrement opalescente, incolore à jaune pâle, et peut contenir des petites particules de protéines translucides ou blanches.
Des instructions complètes pour l'administration sont données dans la notice, à la rubrique 7 « Instructions pour l'utilisation du stylo prérempli d'Erelzi ».
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I
Prescription réservée aux spécialistes et services en DERMATOLOGIE
Prescription réservée aux spécialistes et services en MEDECINE INTERNE
Prescription réservée aux spécialistes et services en PEDIATRIE
Prescription réservée aux spécialistes et services en RHUMATOLOGIE
Médicament d'exception.
Remboursement en fonction de l'indication (JO du 24/11/2017) :
Les seules
indications thérapeutiques ouvrant droit à la prise en charge ou au
remboursement par l'assurance maladie sont, pour les spécialités visées
ci-dessous :
Rhumatisme psoriasique,
Spondylarthrite axiale (spondylarthrite ankylosante et forme non radiographique),
Polyarthrite rhumatoïde modérément à sévèrement active de
l'adulte en cas de réponse inadéquate aux traitements de fond, y
compris le MTX (sauf contre indication),
Psoriasis en plaques chronique sévère, défini par :
. un échec (c'est-à-dire patients non répondeurs, avec une
contre-indication ou intolérants) à au moins deux traitements parmi les
traitements systémiques non biologiques (MTX, ciclosporine et
acitrétine) et la photothérapie et ;
. une surface corporelle atteinte étendue et/ou un retentissement psychosocial important.
- Enfants et adolescents ayant un poids égal ou supérieur à 62,5 kg dans les indications suivantes :
arthrite juvénile idiopathique,
psoriasis en plaques sévère chronique de l'enfant à partir de 6 ans et de l'adolescent.
Solution injectable (injection)
Solution injectable (injection) en stylo prérempli (stylo SensoReady)
La solution est limpide ou légèrement opalescente, et incolore à jaune pâle.
Erelzi 50 mg solution injectable en stylo prérempli
Erelzi est fourni dans une seringue préremplie à usage unique assemblée dans un stylo de forme triangulaire comportant une fenêtre transparente et une étiquette. La seringue se trouvant à l'intérieur du stylo est une seringue transparente en verre transparent (verre de type I) avec une aiguille en acier inoxydable de 29 G x 12,7 mm, un capuchon d'aiguille interne en caoutchouc (élastomère thermoplastique) et un joint de piston en caoutchouc (caoutchouc en bromobutyle), contenant 1,0 mL de solution.
Chaque coffret contient 4 stylos préremplis.
Erelzi 50 mg solution injectable en stylo prérempli
Chaque stylo prérempli contient 50 mg d'étanercept (etanercept).
Étanercept est une protéine de fusion du récepteur p75 du facteur nécrosant des tumeurs. Étanercept est produit par génie génétique et exprimé dans des cellules ovariennes de hamster chinois (CHO).
Pour la liste complète des excipients, voir rubrique Liste des excipients
Acide citrique anhydre
Citrate de sodium dihydraté
Chlorure de sodium
Saccharose
Chlorhydrate de L-lysine
Hydroxyde de sodium (pour l'ajustement du pH)
Acide chlorhydrique (pour l'ajustement du pH)
Eau pour préparations injectables