
NUCALA 100 mg, poudre pour solution injectable, boîte de 1 flacon de 100 mg
Retiré du marché le : 15/05/2024
Dernière révision : 11/10/2022
Taux de TVA : 2.1%
Prix de vente : 917,00 €
Taux remboursement SS : 65%
Base remboursement SS : 917,00 €
Laboratoire exploitant : GLAXOSMITHKLINE
Source :

Asthme sévère à éosinophiles
Nucala est indiqué chez l'adulte, l'adolescent et l'enfant âgé de 6 ans et plus, en traitement additionnel, dans l'asthme sévère réfractaire à éosinophiles (voir rubrique Propriétés pharmacodynamiques).
Polypose naso-sinusienne
Nucala est indiqué en traitement additionnel aux corticostéroïdes par voie nasale chez les patients adultes présentant une polypose naso-sinusienne sévère insuffisamment contrôlés par des corticostéroïdes systémiques et/ou la chirurgie.
Granulomatose éosinophilique avec polyangéite
Nucala est indiqué chez les patients âgés de 6 ans et plus, en traitement additionnel des formes récidivantes ou réfractaires de la granulomatose éosinophilique avec polyangéite.
Syndrome hyperéosinophilique
Nucala est indiqué, en traitement additionnel, chez les patients adultes qui présentent un syndrome hyperéosinophilique insuffisamment contrôlé et sans cause secondaire non hématologique identifiable (voir rubrique Propriétés pharmacodynamiques).
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Traçabilité
Afin d'améliorer la traçabilité des médicaments d'origine biologique, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Exacerbations d'asthme
Le mépolizumab ne doit pas être utilisé pour traiter les exacerbations aiguës d'asthme.
Des symptômes liés à l'asthme ou des exacerbations peuvent survenir pendant le traitement. Les patients doivent prendre un avis médical si leur asthme reste non contrôlé ou s'aggrave après l'instauration du traitement.
Corticoïdes
Il est déconseillé d'arrêter brutalement les corticoïdes après l'instauration du traitement par mépolizumab. Si une réduction des doses de corticoïdes est envisagée, celle-ci doit être progressive et réalisée sous le contrôle d'un médecin.
Réactions d'hypersensibilité et réactions liées à l'administration
Des réactions systémiques immédiates et retardées, incluant des réactions d'hypersensibilité (telles que : anaphylaxie, urticaire, angioœdème, éruption cutanée, bronchospasme, hypotension), ont été observées à la suite de l'administration de mépolizumab. Ces réactions apparaissent généralement dans les heures qui suivent l'administration, mais elles peuvent également survenir plus tardivement (en général après quelques jours). Ces réactions peuvent apparaître pour la première fois alors que le traitement a été initié depuis longtemps (voir rubrique Effets indésirables). En cas de réaction d'hypersensibilité, un traitement approprié adapté au contexte clinique doit être initié.
Parasitoses
Les éosinophiles peuvent être impliqués dans la réponse immunitaire à certaines helminthoses. Les patients présentant une infestation à helminthes doivent être traités avant l'initiation du traitement. En cas d'infestation parasitaire au cours du traitement par mépolizumab et d'échec au traitement antiparasitaire administré, un arrêt temporaire du traitement doit être envisagé.
Granulomatose éosinophilique avec polyangéite menaçant un organe ou le pronostic vital
Nucala n'a pas été étudié chez les patients avec des manifestations de granulomatose éosinophilique avec polyangéite menaçant un organe ou le pronostic vital (voir rubrique Posologie et mode d'administration).
Syndrome hyperéosinophilique menaçant le pronostic vital
Nucala n'a pas été étudié chez les patients avec des manifestations de syndrome hyperéosinophilique menaçant le pronostic vital (voir rubrique Posologie et mode d'administration)
Excipients
Ce médicament contient moins de 1 mmol de sodium (23 mg) pour une dose de 100 mg, c'est-à-dire qu'il est essentiellement « sans sodium ».
Résumé du profil de tolérance
Asthme sévère à éosinophiles
Lors des études contrôlées contre placebo menées chez des patients adultes et adolescents atteints d'asthme sévère réfractaire à éosinophiles, les effets indésirables les plus fréquemment rapportés au cours du traitement ont été : céphalées (20%), réactions au site d'injection (8%) et dorsalgies (6%).
Polypose naso-sinusienne
Lors d'une étude contrôlée contre placebo menée chez des patients atteints de polypose naso- sinusienne, les effets indésirables les plus fréquemment rapportés au cours du traitement ont été : céphalées (18%) et dorsalgies (7%).
Granulomatose éosinophilique avec polyangéite
Lors d'une étude contrôlée contre placebo menée chez des patients atteints de granulomatose éosinophilique avec polyangéite, les effets indésirables les plus fréquemment rapportés au cours du traitement ont été : céphalées (32 %), réactions au site d'injection (15 %) et dorsalgies (13 %). Des réactions d'hypersensibilité/systémiques allergiques ont été rapportées par 4% de patients atteints de granulomatose éosinophilique avec polyangéite.
Syndrome hyperéosinophilique
Lors d'une étude contrôlée contre placebo menée chez des patients atteints de syndrome hyperéosinophilique, les effets indésirables les plus fréquemment rapportés au cours du traitement ont été : céphalées (13%), infections des voies urinaires (9%), réactions au site d'injection et fièvre (7% chacun).
Tableau des effets indésirables
Le tableau ci-dessous présente les effets indésirables observés : chez des patients traités par 100 mg de mépolizumab par voie sous cutanée (SC) (n=263) au cours des études contrôlées contre placebo dans l'asthme sévère à éosinophiles ; chez des patients présentant une polypose naso-sinusienne traités par 100 mg de mépolizumab par voie SC (n=206) au cours d'une étude de 52 semaines randomisée, en double aveugle, contrôlée contre placebo ; chez des patients présentant une granulomatose éosinophilique avec polyangéite (n=68) traités par 300 mg de mépolizumab par voie SC ; chez des patients présentant un syndrome hyperéosinophilique traités par 300 mg de mépolizumab par voie SC (n=54) au cours d'une étude de 32 semaines en double aveugle, contrôlée contre placebo ; et à partir des notifications spontanées de pharmacovigilance depuis la commercialisation. Des données de sécurité sont également disponibles à partir des études d'extension en ouvert chez les patients présentant un asthme sévère réfractaire à éosinophiles (n=998) et traités pendant un temps médian de 2,8 ans (durée de traitement comprise entre 4 semaines et 4,5 ans).
Le profil de sécurité du mépolizumab chez les patients présentant un syndrome hyperéosinophilique (n=102) inclus dans une étude d'extension en ouvert de 20 semaines était similaire au profil de sécurité des patients inclus dans l'étude pivot contrôlée contre placebo.
La fréquence de survenue des effets indésirables est définie selon les critères suivants : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000) ; et fréquence indéterminée (la fréquence de survenue ne peut être estimée sur la base des données disponibles). Dans chaque classe-organe, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
| Classe organe | Effets indésirables | Fréquence |
| Infections et infestations | Infection pulmonaire Infection urinaire Pharyngite | Fréquent |
| Affections du système immunitaire | Réactions d'hypersensibilité (systémiques allergiques)* Anaphylaxie** | Fréquent Rare |
| Affections du système nerveux | Céphalées | Très fréquent |
| Affections respiratoires, thoraciques et médiastinales | Congestion nasale | Fréquent |
| Affections gastro-intestinales | Douleur abdominale haute | Fréquent |
| Affections de la peau et du | Eczéma | Fréquent |
| Classe organe | Effets indésirables | Fréquence |
| tissu sous-cutané | | |
| Affections musculo- squelettiques et systémiques | Dorsalgies | Fréquent |
| Troubles généraux et anomalies au site d'administration | Réactions liées à l'administration (systémiques non-allergiques)*** Réactions locales au site d'injection Fièvre | Fréquent |
* Des réactions systémiques, incluant des réactions d'hypersensibilité, ont été rapportées avec une incidence comparable à celle du placebo dans les études réalisées dans l'asthme sévère à éosinophiles. Voir rubrique Mises en garde spéciales et précautions d'emploi., exemples des manifestations associées ayant été rapportées et leur délai de survenue.
** Effet identifié dans le cadre des déclarations spontanées depuis la commercialisation.
*** Les réactions systémiques non-allergiques liées à l'administration les plus fréquemment rapportées dans les études menées chez les patients présentant un asthme sévère à éosinophiles ont été des éruptions cutanées, des bouffées vaso-motrices et des douleurs musculaires ; ces manifestations ont été rapportées peu fréquemment et chez <1 % des patients ayant reçu 100 mg de mépolizumab par voie sous-cutanée.
Description de certains effets indésirables
Réactions systémiques, incluant des réactions d'hypersensibilité associées à une polypose naso- sinusienne
Dans l'étude de 52 semaines contrôlée contre placebo, 2 patients (<1%) ont rapporté des réactions allergiques (hypersensibilité de type I) systémiques dans le groupe recevant 100 mg de mépolizumab et aucun patient dans le groupe placebo. Aucun patient n'a rapporté d'autres réactions systémiques dans le groupe recevant 100 mg de mépolizumab et 1 patient (<1%) dans le groupe placebo.
Réactions systémiques, incluant les réactions d'hypersensibilité associées à une granulomatose éosinophilique avec polyangéite
Dans l'étude de 52 semaines contrôlée versus placebo, le pourcentage de patients ayant développé des réactions systémiques (allergiques ou non) était de 6 % dans le groupe recevant 300 mg de mépolizumab et de 1 % dans le groupe placebo. Les réactions systémiques, allergiques ou d'hypersensibilité, ont été rapportées chez 4 % des patients dans le groupe recevant 300 mg de mépolizumab et chez 1 % des patients du groupe placebo. Des réactions systémiques non allergiques (angioœdème) ont été rapportées chez 1 (1 %) patient dans le groupe recevant 300 mg de mépolizumab, et chez aucun patient dans le groupe placebo.
Réactions systémiques incluant des réactions d'hypersensibilité associées à un syndrome hyperéosinophilique
Dans l'étude de 32 semaines contrôlée contre placebo, 1 patient (2%) a rapporté une réaction systémique (autre) dans le groupe recevant 300 mg de mépolizumab (réaction cutanée multifocale) et aucun patient dans le groupe placebo.
Réactions locales au site d'injection
Asthme sévère à éosinophiles
Dans les études cliniques contrôlées contre placebo, l'incidence des réactions locales au site d'injection rapportée avec 100 mg de mépolizumab administré par voie sous-cutanée a été de 8 % et 3 % avec le placebo. Tous les évènements rapportés étaient sans gravité, d'intensité légère à modérée et la majorité d'entre eux s'est résorbée en quelques jours. Les réactions locales au site d'injection sont
survenues principalement à l'instauration du traitement et au cours des 3 premières injections ; ces réactions ont été moins fréquemment rapportées au cours des injections suivantes. Les manifestations les plus fréquemment rapportées au cours de ces réactions ont été douleur, érythème, gonflement, démangeaisons et sensation de brûlure.
Polypose naso-sinusienne
Dans l'étude contrôlée contre placebo, des réactions locales au site d'injection (par exemple érythème, prurit) sont survenues chez 2 % des patients recevant 100 mg de mépolizumab, comparé à < 1 % chez les patients recevant le placebo.
Granulomatose éosinophilique avec polyangéite
Dans une étude contrôlée contre placebo, des réactions locales au site d'injection (par exemple : douleur, érythème, gonflement) sont survenues à un taux de 15 % chez les patients recevant 300 mg de mépolizumab, comparé à 13 % chez les patients recevant le placebo.
Syndrome hyperéosinophilique
Dans l'étude contrôlée contre placebo, des réactions locales au site d'injection (par exemple : brulures, démangeaisons) sont survenues à un taux de 7% chez les patients recevant 300 mg de mépolizumab comparé à 4% chez les patients recevant du placebo.
Population pédiatrique
Asthme sévère à éosinophiles
Trente-sept adolescents (âgés de 12-17 ans) ont été inclus dans quatre études d'une durée de 24 à 52 semaines, contrôlées contre placebo (25 patients traités par mépolizumab par voie intraveineuse ou sous-cutanée). Trente-six patients pédiatriques (âgés de 6-11 ans) ont reçu du mépolizumab par voie sous-cutanée pendant 12 semaines dans le cadre d'une étude en ouvert. Après une interruption de traitement de 8 semaines, 30 de ces patients ont reçu du mépolizumab pendant 52 semaines supplémentaires. Le profil de sécurité était similaire à celui observé chez l'adulte. Aucun effet indésirable supplémentaire n'a été identifié.
Syndrome hyperéosinophilique
Quatre adolescents âgés de 12 à 17 ans ont été inclus dans l'étude 200622 contrôlée contre placebo. Un adolescent a reçu 300 mg de mépolizumab et 3 adolescents ont reçu le placebo pendant 32 semaines. Tous les 4 ont poursuivi le traitement dans l'étude d'extension 205203 en ouvert de 20 semaines (voir rubrique Propriétés pharmacodynamiques).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé doivent déclarer tout effet indésirable suspecté via le système national de déclaration - voirAnnexe V.
Les patients présentant une infestation à helminthes doivent être traités avant l'initiation du traitement.
REEVALUER le traitement au minimum une fois par an, en fonction de la gravité de
la maladie du patient et du niveau de contrôle des symptômes.
INFORMER LE MEDECIN si l'asthme reste non contrôlé ou s'aggrave après l'instauration du traitement.
CONSULTER IMMEDIATEMENT LE MEDECIN en cas de :
- Oppression thoracique, toux, difficultés à respirer.
- Evanouissements, vertiges, étourdissements.
- Gonflement au niveau des paupières, du visage, des lèvres, de la langue ou de la bouche.
- Urticaire.
- Eruption cutanée.
Grossesse
Les données sur l'utilisation du mépolizumab chez la femme enceinte sont limitées (moins de 300 grossesses).
Le mépolizumab traverse la barrière placentaire chez les singes. Les études menées chez l'animal n'ont pas mis en évidence de toxicité sur la reproduction (voir rubrique Données de sécurité préclinique). Le risque potentiel pour le fœtus humain n'est pas connu.
Par mesure de précaution, il est préférable d'éviter l'utilisation de Nucala au cours de la grossesse. L'administration de Nucala chez la femme enceinte ne doit être envisagée que si le bénéfice attendu pour la mère est supérieur au risque éventuel pour le fœtus.
Allaitement
Il n'existe pas de donnée sur l'excrétion du mépolizumab dans le lait maternel. Toutefois, le mépolizumab a été retrouvé dans le lait de singes femelles Cynomolgus à des taux inférieurs à 0,5 % des concentrations plasmatiques.
La décision doit être prise, soit d'interrompre l'allaitement, soit d'interrompre le traitement par Nucala, en tenant compte du bénéfice de l'allaitement pour l'enfant et du bénéfice du traitement pour la femme qui allaite.
Fertilité
Il n'existe pas de donnée concernant la fertilité chez les humains. Les études effectuées chez l'animal n'ont pas mis en évidence d'effet indésirable d'un traitement anti-IL-5 sur la fertilité (voir rubrique Données de sécurité préclinique).
Aucune étude d'interaction n'a été réalisée.
Les enzymes du cytochrome P450, les pompes d'efflux et la fixation aux protéines ne sont pas impliqués dans l'élimination du mépolizumab. Une augmentation des concentrations en cytokines pro-inflammatoires (par exemple IL-6) a montré une action inhibitrice sur la formation des enzymes du CYP450 et des transporteurs de médicaments, liée à une interaction avec leurs récepteurs apparentés au niveau des hépatocytes. Néanmoins, l'augmentation des marqueurs pro-inflammatoires systémiques dans l'asthme sévère réfractaire à éosinophiles est faible et aucune expression des récepteurs alpha IL-5 n'a été mise en évidence au niveau des hépatocytes. Par conséquent, le risque d'interactions avec le mépolizumab est considéré comme peu probable.
Nucala doit être prescrit par des médecins expérimentés dans le diagnostic et la prise en charge de l'asthme sévère réfractaire à éosinophiles ou de la polypose naso-sinusienne ou de la granulomatose éosinophilique polyangéite ou du syndrome hyperéosinophilique.
Posologie
Asthme sévère à éosinophiles
Adultes et adolescents âgés de 12 ans et plus
La dose recommandée de mépolizumab est de 100 mg administrés par voie sous-cutanée une fois toutes les 4 semaines.
Enfants âgés de 6 ans à 11 ans
La dose recommandée de mépolizumab est de 40 mg administrés par voie sous-cutanée une fois toutes les 4 semaines.
Nucala est destiné à un traitement au long cours. La nécessité de poursuivre le traitement doit être réévaluée par le médecin au minimum une fois par an, selon un rythme déterminé en fonction de la gravité de la maladie du patient et du niveau de contrôle des exacerbations.
Polypose naso-sinusienne
Adultes
La dose recommandée de mépolizumab est de 100 mg administrés par voie sous-cutanée une fois toutes les 4 semaines.
Nucala est destiné à un traitement au long cours. Des traitements alternatifs peuvent être envisagés chez les patients chez qui aucune réponse au traitement n'est observé après 24 semaines de traitement. Certains patients présentant initialement une réponse partielle peuvent bénéficier d'une amélioration en poursuivant le traitement après 24 semaines.
Granulomatose éosinophilique avec polyangéite
Adultes et adolescents âgés de 12 ans et plus
La dose recommandée de mépolizumab est de 300 mg administrés par voie sous-cutanée une fois toutes les 4 semaines.
La posologie de mépolizumab chez les enfants et les adolescents âgés de 6 à 17 ans atteints de granulomatose éosinophilique avec polyangéite a été déterminée par des modélisations et des données issues de simulation (voir rubrique Propriétés pharmacocinétiques).
Enfants âgés de 6 à 11 ans pesant ≥ 40 kg
La dose recommandée de mépolizumab est de 200 mg, administrés par voie sous-cutanée une fois toutes les 4 semaines.
Enfants âgés de 6 à 11 ans pesant < 40 kg
La dose recommandée de mépolizumab est de 100 mg, administrés par voie sous-cutanée une fois toutes les 4 semaines.
Nucala est destiné à un traitement au long cours. La nécessité de poursuivre le traitement doit être réévaluée par le médecin au minimum une fois par an, selon un rythme déterminé en fonction de la gravité de la maladie et de l'amélioration du contrôle des symptômes.
La nécessité de poursuivre le traitement doit être évaluée chez les patients qui développent des manifestations de granulomatose éosinophilique avec polyangéite menaçant le pronostic vital, compte tenu du fait que Nucala n'a pas été étudié dans cette population.
Syndrome hyperéosinophilique
Adultes
La dose recommandée de mépolizumab est de 300 mg administrés par voie sous-cutanée une fois toutes les 4 semaines.
Nucala est destiné à un traitement au long cours. La nécessité de poursuivre le traitement doit être réévaluée par le médecin au minimum une fois par an, selon un rythme déterminé en fonction de la gravité de la maladie du patient et du niveau de contrôle des symptômes.
La nécessité de poursuivre le traitement doit être évaluée chez les patients qui développent des manifestations de syndrome hyperéosinophilique menaçant le pronostic vital, compte tenu du fait que Nucala n'a pas été étudié dans cette population.
Populations spécifiques
Sujets âgés
Aucune adaptation posologique n'est nécessaire chez les personnes âgées (voir rubrique Propriétés pharmacocinétiques).
Insuffisance rénale et hépatique
Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance rénale ou hépatique (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
Asthme sévère à éosinophiles
Enfants âgés de moins de 6 ans
La sécurité et l'efficacité du mépolizumab chez les enfants âgés de moins de 6 ans n'ont pas encore été établies.
Aucune donnée n'est disponible. Enfants âgés de 6 à 17 ans
La posologie du mépolizumab chez l'enfant et l'adolescent (âgés de 6 ans à 17 ans) présentant un asthme sévère réfractaire à éosinophiles a été déterminée sur la base de données limitées issues d'études d'efficacité, de pharmacocinétique et de pharmacodynamie ainsi que de résultats obtenus à partir de modélisation et de simulation (voir rubriques Propriétés pharmacodynamiques et Propriétés pharmacocinétiques).
Polypose naso-sinusienne chez les enfants âgés de moins de 18 ans
La sécurité et l'efficacité du mépolizumab chez les enfants âgés de moins de 18 ans présentant une polypose naso-sinusienne n'ont pas été établies. Aucune donnée n'est disponible.
Granulomatose éosinophilique avec polyangéite chez les enfants âgés de moins de 6 ans
La sécurité et l'efficacité de mépolizumab n'ont pas été établies chez les enfants de moins de 6 ans. Aucune donnée n'est disponible.
Syndrome hyperéosinophilique chez les enfants âgés de moins de 18 ans
La sécurité et l'efficacité du mépolizumab chez les enfants et les adolescents âgés de moins de 18 ans n'ont pas encore été établis.
Les données disponibles actuellement sont décrites en rubriques Effets indésirables, Propriétés pharmacodynamiques et Propriétés pharmacocinétiques mais aucune recommandation de posologie ne peut être faite.
Mode d'administration
Nucala doit être administré exclusivement par injection sous-cutanée par un professionnel de santé. L'injection peut se faire, soit dans la partie supérieure du bras, soit au niveau de la cuisse ou de l'abdomen.
Dans le cas d'une dose nécessitant plus d'une injection, il est recommandé que les injections soient espacées d'au moins 5 cm.
Avant administration, la solution injectable sera reconstituée à partir de la poudre. La solution reconstituée doit être utilisée immédiatement. Voir rubrique Précautions particulières d'élimination et de manipulation pour les instructions concernant la reconstitution du médicament avant administration.
Chaque flacon de mépolizumab doit être destiné à un patient unique, et toute solution non utilisée doit être jetée.
Durée de conservation :
4 ans.
Après reconstitution
La stabilité physico-chimique de la solution reconstituée a été démontrée pendant 8 heures lorsqu'elle est conservée à une température inférieure à 30 °C.
D'un point de vue microbiologique, la solution reconstituée doit être utilisée immédiatement, à moins que les modalités de reconstitution excluent totalement tout risque de contamination microbiologique. Si elle n'est pas utilisée immédiatement, la durée et les conditions de conservation sont sous la responsabilité de l'utilisateur.
Précautions particulières de conservation :
A conserver à une température ne dépassant pas 25°C. Ne pas congeler.
Conserver le flacon dans l'emballage extérieur, à l'abri de la lumière.
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique Durée de conservation.
Ce médicament ne doit pas être mélangé avec d'autres médicaments.
Dans un essai clinique, des doses uniques allant jusqu'à 1 500 mg ont été administrées par voie intraveineuse à des patients atteints de syndrome hyperéosinophilique sans que soit mis en évidence une toxicité dose-dépendante.
Il n'existe pas de traitement spécifique en cas de surdosage en mépolizumab. En cas de surdosage, le patient doit recevoir un traitement symptomatique et être placé sous surveillance si nécessaire.
Une prise en charge complémentaire sera instaurée si elle est médicalement indiquée ou recommandée par les Centres Antipoison nationaux, le cas échéant.
Classe pharmacothérapeutique : Médicaments pour les maladies obstructives des voies respiratoires, autres médicaments systémiques pour les maladies obstructives des voies respiratoires, Code ATC : R03DX09.
Mécanisme d'action
Le mépolizumab est un anticorps monoclonal humanisé (IgG1, kappa), qui cible l'interleukine-5 (IL-5) humaine avec une affinité et une spécificité élevées. L'IL-5 est la principale cytokine intervenant dans la croissance et la différenciation, le recrutement, l'activation et la durée de vie des éosinophiles. Le mépolizumab inhibe l'activité biologique de l'IL-5 à une concentration nanomolaire en bloquant la liaison de l'IL-5 à la chaîne alpha du complexe récepteur de l'IL-5 exprimé sur la surface cellulaire des éosinophiles. Il inhibe ainsi la voie de signalisation de l'IL-5 et réduit la production et la durée de vie des éosinophiles.
Effets pharmacodynamiques
Asthme sévère à éosinophiles
Chez les patients présentant un asthme sévère réfractaire à éosinophiles (adultes/adolescents), après l'administration par voie sous-cutanée d'une dose de 100 mg de mépolizumab toutes les 4 semaines pendant 32 semaines, le taux moyen (moyenne géométrique) d'éosinophiles sanguins a été réduit de 290 cellules/µl à l'inclusion à 40 cellules/µl à la semaine 32 (n=182) ; avec une réduction de 84 % par rapport au placebo. L'amplitude de cette réduction d'éosinophiles sanguins s'est maintenue chez les patients présentant un asthme sévère réfractaire à éosinophiles (n=998) et traités pendant un temps médian de 2,8 ans (durée de traitement comprise entre 4 semaines et 4,5 ans) dans les études d'extension en ouvert.
Chez des enfants âgés de 6 ans à 11 ans, présentant un asthme sévère réfractaire à éosinophiles et recevant mépolizumab par voie sous-cutanée toutes les 4 semaines pendant 52 semaines, la moyenne géométrique des taux d'éosinophiles sanguins est passée de 306 (n=16) (à l'inclusion) à 48 cellules/µL (semaine 52) (n=15) après administration de 40 mg (pour un poids < 40 kg) et de 331 à 44 cellules/µL (n=10) après administration de 100 mg (pour un poids ≥ 40 kg), avec une réduction par rapport aux taux à l'inclusion de respectivement 85 % et 87 %.
Chez l'adulte, l'adolescent et l'enfant, l'intensité de l'effet a été maintenue durant les 4 semaines du traitement.
Polypose naso-sinusienne
Chez les patients présentant une polypose naso-sinusienne, après l'administration d'une dose de 100 mg de mépolizumab par voie sous-cutanée toutes les 4 semaines pendant 52 semaines, les éosinophiles sanguins ont été réduits : la moyenne géométrique des taux d'éosinophiles sanguins passant de 390 cellules/µL à l'inclusion (n=206) à 60 cellules/µL (n=126) à la semaine 52, ce qui correspond à une réduction de la moyenne géométrique de 83% comparé au placebo. L'intensité de la réduction a été observée durant les 4 semaines de traitement et a été maintenue tout au long de la période de traitement de 52 semaines.
Granulomatose éosinophilique avec polyangéite
Chez les patients présentant une granulomatose éosinophilique avec polyangéite, après administration d'une dose de 300 mg de mépolizumab par voie sous-cutanée toutes les 4 semaines pendant 52 semaines, la moyenne géométrique du taux d'éosinophiles sanguins a diminué, passant de 177 (n=68) à l'inclusion à 38 cellules/µL (n=64) à la semaine 52. Une réduction de la moyenne géométrique de 83 % comparé au placebo a été observée. L'intensité de cette réduction a été maintenue tout au long des 4 semaines de traitement.
Syndrome hyperéosinophilique
Chez les patients présentant un syndrome hyperéosinophilique (adultes et adolescents), après administration d'une dose de 300 mg de mépolizumab par voie sous-cutanée toutes les 4 semaines pendant 32 semaines, la réduction des éosinophiles sanguins a été observée après deux semaines de traitement. A la semaine 32, la moyenne géométrique du taux d'éosinophiles sanguins a diminué, passant de 1460 à l'inclusion (n=54) à 70 cellules/µL (n=48) et une réduction de la moyenne géométrique de 92% comparé au placebo a été observée. L'intensité de la réduction a été maintenue pendant 20 semaines supplémentaires chez les patients qui avaient continué le traitement par mépolizumab dans l'étude d'extension en ouvert.
Immunogénicité
Asthme sévère à éosinophiles, polypose naso-sinusienne, granulomatose éosinophilique avec polyangéite et syndrome hyperéosinophilique
Compte-tenu des propriétés potentiellement immunogènes des protéines et peptides utilisés en thérapeutique, des anticorps dirigés contre le mépolizumab peuvent apparaître suite au traitement. Dans les études cliniques contrôlées contre placebo, après avoir reçu au moins une dose de mépolizumab par voie sous-cutanée, des taux d'anticorps anti-mépolizumab détectables ont été observés chez 15/260 (6%) adultes et adolescents présentant un asthme sévère à éosinophiles traités à la dose de 100 mg, chez 6/196 (3%) adultes présentant une polypose naso-sinusienne traités à la dose de 100 mg, chez 1/68 (<2%) adultes présentant une granulomatose éosinophilique avec polyangéite traités à la dose de 300 mg et chez 1/53 (2%) adultes et adolescents présentant un syndrome hyperéosinophilique traités à la dose de 300 mg.
Le profil d'immunogénicité du mépolizumab chez les patients présentant un asthme sévère réfractaire à éosinophiles (n=998) traités pendant un temps médian de 2,8 ans (durée de traitement comprise entre 4 semaines et 4,5 ans) ou chez les patients présentant un syndrome hyperéosinophilique (n=102) traités pendant 20 semaines dans les études d'extension en ouvert était similaire à celui observé dans les études contrôlées contre placebo.
Chez les enfants âgés de 6 ans à 11 ans, présentant un asthme sévère réfractaire à éosinophiles, des taux d'anticorps anti-mépolizumab ont été détectés chez 2/35 (6 %) d'entre eux après administration de 40 mg (pour un poids < 40 kg) ou de 100 mg (pour un poids ≥ 40 kg) de mépolizumab et ce, après administration d'au moins une dose de mépolizumab au cours de la phase d'initiation de courte durée de l'étude. Aucun enfant n'a développé d'anticorps anti-mépolizumab pendant toute la phase à long terme de l'étude.
Des anticorps neutralisants ont été détectés chez un patient adulte présentant un asthme sévère réfractaire à éosinophiles. Il n'a pas été observé d'anticorps neutralisants chez les patients présentant une polypose naso-sinusienne, une granulomatose éosinophilique avec polyangéite ou un syndrome hyperéosinophilique. Les anticorps anti-mépolizumab n'ont pas eu d'impact notable sur les paramètres pharmacocinétiques et pharmacodynamiques du traitement chez la majorité des patients. Il n'a pas été établi de corrélation entre les taux d'anticorps et la variation du nombre d'éosinophiles circulants.
Efficacité clinique
Asthme sévère à éosinophiles
L'efficacité du mépolizumab a été évaluée dans la population cible des patients présentant un asthme sévère réfractaire à éosinophiles dans 3 études cliniques randomisées, en double aveugle et en groupes parallèles, conduites sur une période allant de 24 à 52 semaines et incluant des patients âgés d'au moins 12 ans. Ces patients présentaient soit un asthme dont les symptômes n'étaient pas contrôlés (au moins deux exacerbations d'asthme au cours de l'année précédente) par leur traitement standard en cours, incluant au minimum des corticoïdes inhalés (CSI) à fortes doses et un ou plusieurs autre(s) traitement(s) de fond de l'asthme, soit un asthme corticodépendant. Les traitements de fond additionnels comprenaient les bêta2-agonistes de longue durée d'action (LABA), les antileucotriènes, les antagonistes muscariniques de longue durée d'action (LAMA), la théophylline et les corticoïdes oraux.
Les deux études ayant évalué l'effet du traitement sur les exacerbations, MEA112997 et MEA115588, ont inclus un total de 1 192 patients, âgés de 49 ans en moyenne (allant de 12 à 82 ans), dont 60 % de femmes. La proportion de patients recevant des corticoïdes oraux en traitement de fond a été respectivement dans chaque étude de 31 % et 24 %. A l'inclusion, les patients devaient avoir eu au moins 2 épisodes d'exacerbation de l'asthme sévère au cours des 12 derniers mois nécessitant de recourir à un traitement par corticoïdes oraux ou systémiques et une fonction pulmonaire altérée (VEMS pré-bronchodilatateur < 80 % chez l'adulte et < 90 % chez l'adolescent). Le nombre moyen d'exacerbations rapportées au cours des 12 derniers mois était de 3,6 et le VEMS pré- bronchodilatateur moyen à l'inclusion était de 60 %. Les patients ont poursuivi leur traitement habituel de l'asthme tout au long des études.
L'étude ayant évalué la réduction des doses de corticoïdes oraux, MEA115575, a inclus un total de 135 patients, traités quotidiennement par des corticoïdes oraux (de 5 à 35 mg par jour) et des corticoïdes inhalés à fortes doses associés à un autre traitement de fond (55 % de femmes ; âge moyen 50 ans).
Étude de dose-efficacité MEA112997 (DREAM)
L'étude MEA112997, étude multicentrique randomisée, en double aveugle, contrôlée contre placebo et en groupes parallèles, a été conduite sur 52 semaines chez 616 patients atteints d'asthme sévère réfractaire à éosinophiles. Le mépolizumab était administré par voie intraveineuse aux doses de 75 mg, 250 mg ou 750 mg. Une réduction significative des exacerbations cliniquement significatives de l'asthme (définies comme une aggravation de l'asthme imposant l'utilisation de corticoïdes oraux/systémiques et/ou une hospitalisation et/ou une consultation aux urgences) a été observée par rapport au placebo (voir le tableau 1).
Tableau 1 : Fréquence des exacerbations cliniquement significatives à la semaine 52 (analyse en intention de traiter)
| | Mépolizumab par voie intraveineuse | Placebo | ||
| 75 mg n = 153 | 250 mg n = 152 | 750 mg n = 156 | n = 155 | |
| Taux annuel d'exacerbations | 1,24 | 1,46 | 1,15 | 2,40 |
| Pourcentage de réduction | 48 % | 39 % | 52 % | |
| Ratio des taux (IC à 95 %) | 0,52 (0,39 - 0,69) | 0,61 (0,46 - 0,81) | 0,48 (0,36 - 0,64) | |
| Valeur de p | < 0,001 | < 0,001 | < 0,001 | - |
Étude MEA115588 sur la réduction des exacerbations (MENSA)
L'étude MEA115588 était une étude multicentrique randomisée, en double aveugle, contrôlée versus placebo et en groupes parallèles, qui a évalué l'efficacité et la tolérance du mépolizumab en traitement additionnel chez 576 patients atteints d'asthme sévère réfractaire à éosinophiles défini par un taux d'éosinophiles dans le sang périphérique supérieur ou égal à 150 cellules/µl à l'instauration du traitement ou supérieur ou égal à 300 cellules/µl au cours des 12 derniers mois.
Les traitements étudiés étaient 100 mg de mépolizumab par voie sous-cutanée, 75 mg de mépolizumab par voie intraveineuse ou le placebo, administrés une fois toutes les 4 semaines pendant 32 semaines. Le critère d'évaluation principal était la fréquence des exacerbations de l'asthme cliniquement significatives. Une réduction statistiquement significative de la fréquence des exacerbations cliniquement significatives a été observée dans les deux bras de traitement mépolizumab par comparaison au placebo (p < 0,001). Le tableau 2 présente les résultats du critère d'évaluation principal et des critères d'évaluation secondaires dans le groupe traité par le mépolizumab par voie sous-cutanée et dans le groupe placebo.
Tableau 2 : Résultats du critère d'évaluation principal et des critères d'évaluation secondaires à la semaine 32 dans la population en intention de traiter (MEA115588)
| | Mépolizumab | Placebo |
| 100 mg | | |
| (voie sous-cutanée) | n = 191 | |
| n = 194 | | |
| Critère d'évaluation principal | ||
| Fréquence des exacerbations cliniquement significatives | ||
| Taux annuel d'exacerbations | 0,83 | 1,74 |
| Pourcentage de réduction | 53 % | - |
| Ratio des taux (IC à 95 %) | 0,47 (0,35 - 0,64) | |
| Valeur de p | < 0,001 | |
| Critères d'évaluation secondaires | ||
| Fréquence des exacerbations nécessitant des hospitalisations/consultations aux urgences | ||
| Taux annuel d'exacerbations | 0,08 | 0,20 |
| Pourcentage de réduction | 61 % | _ |
| Ratio des taux (IC à 95 %) | 0,39 (0,18 - 0,83) | |
| Valeur de p | 0,015 | |
| Fréquence des exacerbations nécessitant une hospitalisation | ||
| Taux annuel d'exacerbations | 0,03 | 0,10 |
| Pourcentage de réduction | 69 % | _ |
| Ratio des taux (IC à 95 %) | 0,31 (0,11 - 0,91) | |
| Valeur de p | 0,034 | |
| VEMS pré-bronchodilatateur (ml) à la semaine 32 | ||
| Valeur à l'inclusion (écart- type) | 1 730 (659) | 1 860 (631) |
| Changement moyen par rapport à la valeur initiale (écart-type) | 183 (31) | 86 (31) |
| Différence (mépolizumab vs placebo) | 98 | |
| IC à 95 % | (11 - 184) | |
| Valeur de p | 0,028 | |
| Score du questionnaire respiratoire de Saint George (SGRQ) à la semaine 32 | ||
| Valeur à l'inclusion (écart- type) | 47,9 (19,5) | 46,9 (19,8) |
| Changement moyen du score par rapport à la valeur initiale (écart-type) | -16,0 (1,1) | -9,0 (1,2) |
| Différence (mépolizumab vs placebo) | -7,0 | |
| IC à 95 % | (-10,2 - -3,8) | |
| Valeur de p | < 0,001 | |
Réduction du taux d'exacerbations en fonction du taux d'éosinophiles sanguins à l'inclusion
Le tableau 3 présente les résultats d'une analyse groupée des deux études évaluant l'effet sur les exacerbations (MEA112997 et MEA115588) en fonction du taux d'éosinophiles sanguins à l'inclusion. Le taux d'exacerbations dans le groupe placebo a augmenté avec l'augmentation du taux d'éosinophiles sanguins à l'inclusion. Avec le mépolizumab, la réduction du taux d'exacerbations a été plus importante chez les patients ayant un taux d'éosinophiles sanguins plus élevé.
Table 3 : Analyse groupée du taux d'exacerbations cliniquement significatives en fonction du taux d'éosinophiles sanguins à l'inclusion chez les patients ayant un asthme sévère réfractaire à éosinophiles
| | Mépolizumab 75 mg IV/100 mg SC n=538 | Placebo n=346 |
| MEA112997+MEA115588 | | |
| <150 cellules/µl | ||
| n | 123 | 66 |
| Taux annuel d'exacerbations | 1,16 | 1,73 |
| Mépolizumab vs. Placebo | | |
| Ratio des taux (IC à 95 %) | 0,67 (0,46 - 0,98) | --- |
| 150 à <300 cellules/µl | ||
| n | 139 | 86 |
| Taux annuel d'exacerbations | 1,01 | 1,41 |
| Mépolizumab vs. Placebo | | |
| Ratio des taux (IC à 95 %) | 0,72 (0,47 - 1,10) | --- |
| 300 à <500 cellules/µl | ||
| n | 109 | 76 |
| Taux annuel d'exacerbations | 1,02 | 1,64 |
| Mépolizumab vs. Placebo | | |
| Ratio des taux (IC à 95 %) | 0,62 (0,41 - 0,93) | --- |
| ≥500 cells/µL | ||
| n | 162 | 116 |
| Taux annuel d'exacerbations | 0,67 | 2,49 |
| Mépolizumab vs. Placebo | | |
| Ratio des taux (IC à 95 %) | 0,27 (0,19 - 0,37) | --- |
Étude sur la réduction des corticoïdes oraux MEA115575 (SIRIUS)
L'étude MEA115575 a évalué chez des patients présentant un asthme sévère réfractaire à éosinophiles, l'effet de 100 mg de mépolizumab administré par voie sous-cutanée sur la réduction du besoin de recourir à un traitement de fond par des corticoïdes oraux, tout en maintenant le contrôle de l'asthme. Les patients présentaient un taux d'éosinophiles sanguins ≥ 150/µl à l'inclusion ou ≥ 300/?l dans les 12 mois précédant la sélection. Les patients ont reçu soit un traitement par mépolizumab, soit par placebo, une fois toutes les 4 semaines pendant la période de traitement. Les patients ont poursuivi leur traitement préexistant de l'asthme au cours de l'étude, à l'exception de leur dose de corticoïdes oraux qui était réduite progressivement toutes les 4 semaines pendant la phase de réduction des corticoïdes oraux (semaines 4 à 20) et ceci tant que l'asthme restait contrôlé.
Un total de 135 patients a été inclus (âge moyen 50 ans, 55 % de femmes) dont 48 % avaient reçu une corticothérapie orale pendant au moins 5 ans. La dose moyenne de prednisone (ou équivalent) à l'inclusion était d'environ 13 mg par jour.
Le critère d'évaluation principal était le pourcentage de réduction de la dose quotidienne de corticoïdes oraux (semaines 20 à 24) atteint tout en maintenant le contrôle de l'asthme selon des catégories de réduction de doses définies (voir le tableau 4). Les catégories ont été prédéfinies par des intervalles de pourcentage de réduction allant de 90-100 % de réduction jusqu'à aucune réduction de la dose de prednisone, à partir de la fin de la phase d'optimisation. La différence entre le mépolizumab et le placebo a été statistiquement significative (p=0,008).
Tableau 4 : Résultats du critère d'évaluation principal et les critères d'évaluation secondaires de l'étude MEA115575
| | Population en intention de traiter | |
| | Mépolizumab 100 mg (voie sous-cutanée) n = 69 | Placebo n = 66 |
| Critère d'évaluation principal | ||
| Pourcentage de réduction de la dose des corticoïdes oraux par rapport à la valeur initiale (semaines 20 à 24) | 16 (23 %) 12 (17 %) 9 (13 %) 7 (10 %) 25 (36 %) | 7 (11 %) 5 (8 %) 10 (15 %) 7 (11 %) 37 (56 %) |
| 90
% à 100 % 75 % à < 90 % 50 % à < 75 % > 0 % à < 50 % Aucune réduction des corticoïdes oraux / asthme non contrôlé / arrêt du traitement | ||
| Odds ratio (IC à 95 %) | 2,39 (1,25 - 4,56) | |
| Valeur de p | 0,008 | |
| Critères d'évaluation secondaires (semaines 20 à 24) | ||
| Réduction de la dose quotidienne de corticoïdes oraux jusqu'à 0 mg/jour | 10 (14 %) | 5 (8 %) |
| Odds ratio (IC à 95 %) | 1,67 (0,49 - 5,75) | |
| Valeur de p | 0,414 | |
| Réduction de la dose quotidienne de corticoïdes oraux jusqu'à ≤ 5 mg/jour | 37 (54 %) | 21 (32 %) |
| Odds ratio (IC à 95 %) | 2,45 (1,12 - 5,37) | |
| Valeur de p | 0,025 | |
| % médian de réduction de la dose quotidienne de corticoïdes oraux par rapport à la valeur initiale (IC à 95 %) | 50,0 (20,0 - 75,0) | 0,0 (-20,0 - 33,3) |
| Différence médiane (IC à95 %) | -30,0 (-66,7 - 0,0) | |
| Valeur de p | 0,007 | |
Etudes d'extension en ouvert chez les patients atteints d'asthme sévère réfractaire à éosinophiles : MEA115666 (COLUMBA), MEA115661 (COSMOS) et 201312 (COSMEX)
Le profil d'efficacité à long-terme du mépolizumab chez les patients présentant un asthme sévère réfractaire à éosinophiles (n=998) et traités pendant un temps médian de 2,8 ans (durée de traitement comprise entre 4 semaines et 4,5 ans) dans les études d'extension en ouvert MEA115666, MEA115661 et 201312 était généralement cohérent par rapport à celui observé dans les 3 études contrôlées contre placebo.
Polypose naso-sinusienne
L'étude 205687 (SYNAPSE) était une étude de 52 semaines, randomisée, en double aveugle, contrôlée contre placebo, qui a évalué 407 patients âgés de 18 ans et plus présentant une polypose naso-sinusienne.
Les patients inclus dans l'étude devaient avoir un score de symptômes d'obstruction nasale selon l'échelle visuelle analogique (EVA) >5 sur un score maximal de 10, un score de symptôme global >7 sur un score maximal de 10 et un score endoscopique bilatéral de polypose nasale ≥ 5 sur un score maximal de 8 (avec un score minimal de 2 dans chaque narine). Les patients devaient également avoir eu au moins une chirurgie des polypes nasaux au cours des 10 dernières années.
Les principales caractéristiques à l'inclusion comprenaient : score total moyen endoscopique de polypose nasale de 5,5 (ET=1,29) ; score d'obstruction nasale moyen selon l'EVA de 9,0 (ET = 0,83) ; score global moyen des symptômes selon l'EVA de 9,1 (ET = 0,74) ; score moyen de la perte d'odorat de 9,7 (ET = 0,72) ; score total moyen SNOT-22 de 64,1 (ET = 18,32). La moyenne géométrique du taux d'éosinophiles était de 390 cellules/µL (IC 95% : 360 ; 420). De plus, 27 % des patients présentaient une Maladie Respiratoire Aggravée par l'Aspirine et 48% avaient eu au moins un traitement par corticostéroïdes oraux pour la polypose naso-sinusienne au cours des 12 derniers mois.
Les patients ont reçu une dose de 100 mg de mépolizumab ou de placebo, administrée par voie sous- cutanée une fois toutes les 4 semaines, en plus de leur traitement de fond par corticothérapie intranasale.
Les co-critères d'évaluation principaux étaient la variation du score endoscopique total de polypose nasale entre l'inclusion dans l'étude et la semaine 52 et la variation de la moyenne du score d'obstruction nasale selon l'EVA entre l'inclusion et les semaines 49 à 52. Le critère d'évaluation secondaire clé était le délai avant la première chirurgie des polypes nasaux pendant l'étude jusqu'à la semaine 52 (la chirurgie était définie comme toute intervention entraînant une incision et une ablation de tissu [par exemple, une polypectomie] dans la cavité nasale). Les patients qui ont reçu le mépolizumab ont présenté des améliorations (diminutions) significativement plus importantes du score total endoscopique de PN à la semaine 52 et du score d'obstruction nasale selon l'EVA au cours des semaines 49 à 52 par rapport au placebo, et tous les critères d'évaluation secondaires étaient statistiquement significatifs en faveur du mépolizumab (voir Tableau 5 et Figure 1).
Tableau 5 : Résumé des résultats des critères primaires et secondaires (population en intention de traiter)
| | Placebo (N=201) | Mepolizumab 100 mg SC (N=206) |
| Co-critères d'évaluation | | |
| Score total endoscopique à la semaine 52 a | | |
| Score médian à l'inclusion dans l'étude (min ; max) | 6.0 (0 ; 8) | 5,0 (2 ; 8) |
| Variation médiane par rapport à l'inclusion dans l'étude | 0.0 | -1,0 |
| Valeur de p b | | <0,001 |
| Différence de médianes (IC 95%) c | | -0,73 (-1,11 ; -0,34) |
| Amélioration ≥1-point, n (%) | 57 (28) | 104 (50) |
| Amélioration ≥2-point, n (%) | 26 (13) | 74 (36) |
| Score d'obstruction nasale selon l'EVA (semaines 49 à 52) a | | |
| Score médian à l'inclusion dans l'étude (min ; max) | 9.14 (5,31 ; 10,00) | 9.01 (6.54 ; 10,00) |
| Variation médiane par rapport à l'inclusion dans l'étude | -0,82 | -4,41 |
| Valeur de p b | | <0,001 |
| Différence de médianes (IC 95%) c | | -3,14 (-4,09 ; -2,18) |
| Amélioration ≥1-point, n (%) | 100 (50) | 146 (71) |
| Amélioration ≥3-point, n (%)d | 73 (36) | (60) |
| Critère secondaire clé | | |
| Délai avant la première chirurgie des polypes nasaux | | |
| Participants avec chirurgie | 46 (23) | 18 (9) |
| Hazard ratio (Mepolizumab/Placebo) (IC 95%) e | | 0,43 (0,25 ; 0,76) |
| Valeur de pe | | 0,003 |
| Autres critères d'évaluation secondaires | | |
| Score total selon l'EVA (semaines 49 à 52) a | ||
| Score médian à l'inclusion dans l'étude (min ; max) | 9,20 (7,21 ; 10,00) | 9,12 (7,17 ; 10,00) |
| Variation médiane par rapport à l'inclusion dans l'étude | -0,90 | -4,48 |
| Valeur de p b | | <0,001 |
| Différence de médianes (IC 95%) c | | -3,18 (-4,10 ; -2,26) |
| Amélioration ≥ 2,5-point (%)f | 40 | 64 |
| Score total SNOT-22 à la semaine 52 a, g | ||
| n | 198 | 205 |
| Score médian à l'inclusion dans l'étude (min, max) | 64,0 (19 ; 110) | 64,0 (17 ; 105) |
| Variation médiane par rapport à l'inclusion dans l'étude | -14,0 | -30,0 |
| Valeur de p b | | <0,001 |
| Différence de médianes (IC 95%) c | | -16,49 (-23,57 ; -9,42) |
| Amélioration ≥28-point (%)f | 32 | 54 |
| Patients ayant eu recours à des corticostéroïdes systémiques pour des polypes nasaux jusqu'à la semaine 52 | ||
| Nombre de patient avec ≥1 traitement | 74 (37) | 52 (25) |
| Odds Ratio par rapport au Placebo (IC 95%)h | 0,58 (0,36 ; 0,92) | |
| Valeur de ph | 0,020 | |
| Score composite selon l'EVA - Symptômes nasaux (semaines 49 à 52) a, i | ||
| Score médian à l'inclusion dans l'étude (min ; max) | 9,18 (6.03 ; 10.00) | 9,11 (4,91 ; 10,00) |
| Variation médiane par rapport à l'inclusion dans l'étude | -0.89 | -3,96 |
| Valeur de p b | | <0,001 |
| Différence de médianes (95% IC) c | | -2,68 (-3,44 ; -1,91) |
| Amélioration ≥2-point (%)f | 40 | 66 |
| Score de perte de l'odorat selon l'EVA (semaines 49 à 52) a | ||
| Score médian à l'inclusion dans l'étude (min ; max) | 9,97 (6,69 ; 10,00) | 9,97 (0,94 ; 10,00) |
| Variation médiane par rapport à l'inclusion dans l'étude | 0,00 | -0,53 |
| Valeur de p b | | <0,001 |
| Différence de médianes (IC 95%) c | | -0,37 (-0,65 ; -0,08) |
| Amélioration ≥3-point (%)f | 19 | 36 |
a Les patients ayant subi une chirurgie nasale/sinusoplastie avant la visite se sont vus attribuer leur plus mauvais score observé avant leur chirurgie nasale/sinusoplastie. Les patients qui ont arrêtés l'étude sans avoir subi de chirurgie nasale/sinusoplastie se sont vu attribuer leur plus mauvais score observé avant leur retrait de l'étude.
b Basé sur le test de la somme des rangs de Wilcoxon
c Régression quantile avec comme co-variables : groupe de traitement, région géographique, score à l'inclusion et log(e) du taux d'éosinophiles à l'inclusion.
d Une amélioration de trois points de l'obstruction nasale selon l'EVA a été identifiée comme étant un changement significatif pour le patient pour cette évaluation.
e Estimé selon un modèle de Cox à risque proportionnel avec le groupe de traitement, la région géographique, le score total endoscopique à l'inclusion (lu au niveau central), l'obstruction nasale à l'inclusion selon l'EVA, le log(e) du taux d'éosinophiles à l'inclusion et le nombre de chirurgies précédentes (1, 2, >2 en ordinal) comme co-variables.
f Le seuil d'amélioration a été identifié comme étant un changement significatif pour le patient pour cette évaluation.
g Amélioration observée dans les 6 domaines des symptômes et conséquences associés à la polypose naso-sinusienne.
h Analyse utilisant un modèle de régression logistique avec comme co-variables : groupe de traitement, région géographique, nombre de traitement par corticostéroïdes oraux pour les polypes nasaux au cours des 12 derniers mois (0, 1, >1 en ordinal), score endoscopique total de polypose nasale à l'inclusion (lu au niveau central), score d'obstruction nasale selon l'EVA à l'inclusion et log(e) du taux d'éosinophiles à l'inclusion.
i Score composite selon l'EVA incluant l'obstruction nasale, l'écoulement nasal, les mucosités dans la gorge et la perte d'odorat.
Délai avant la première chirurgie des polypes nasaux pendant l'étude
Au cours de la période de traitement de 52 semaines, les patients du groupe mépolizumab avaient une probabilité plus faible de recourir à une chirurgie des polypes nasaux que les patients du groupe placebo. Le risque de chirurgie sur la période de traitement était significativement plus faible de 57% pour les patients traités par mépolizumab par rapport au placebo (Hazard ratio : 0,43 ; IC 95% 0,25 ; 0,76 ; p=0,003).
Figure 1: Courbe de Kaplan Meier du délai avant la première chirurgie des polypes nasaux
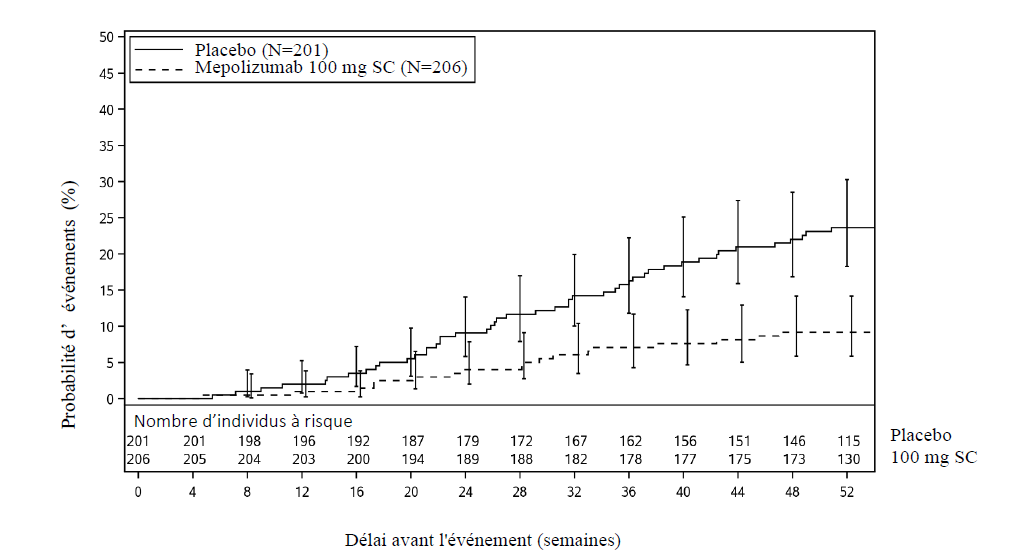
Une analyse post-hoc de la proportion de patients avec chirurgie a montré une réduction de 61 % de la probabilité de chirurgie par rapport au placebo (OR : 0,39, IC 95 % : 0,21 ; 0,72 ; p= 0,003).
Patients présentant une polypose naso-sinusienne avec un asthme associé
Chez 289 (71%) patients avec un asthme associé, les analyses pré-définies ont montré des améliorations des co-critères d'évaluation principaux cohérentes avec celles observées dans la population globale chez les patients ayant reçu 100 mg de mépolizumab par rapport au placebo. De plus, chez ces patients, il a été observé une amélioration plus importante entre l'inclusion dans l'étude et la semaine 52 du contrôle de l'asthme mesuré par le Questionnaire de Contrôle de l'Asthme (ACQ- 5) pour le mépolizumab 100 mg par rapport au placebo (changement médian [Q1, Q3] de -0,80 [-2,20, 0,00] et 0,00 [-1,10 ; 0,20], respectivement).
Granulomatose éosinophilique avec polyangéite
L'étude MEA115921, une étude randomisée de 52 semaines, en double aveugle, contrôlée contre placebo, a évalué 136 patients adultes atteints de granulomatose éosinophilique avec polyangéite avec antécédents de forme de la maladie récidivante ou réfractaire et recevant une dose stable de corticothérapie orale (corticostéroïdes par voie orale ; ≥ 7,5 à ≤50 mg/jour de prednisolone/prednisone), avec ou sans traitement immunosuppresseur avec dose stable (à l'exclusion de la cyclophosphamide). D'autres traitements de fond standards étaient autorisés pendant l'étude. 53% (n=72) des patients recevaient de façon concomitante un traitement immunosuppresseur avec dose stable. Les patients présentant une granulomatose éosinophilique avec polyangéite menaçant un organe ou le pronostic vital ont été exclus de l'étude MEA115921.
Les patients ont reçu soit 300 mg de mépolizumab soit un placebo, administrés par voie sous-cutanée une fois toutes les 4 semaines, en plus de leur traitement de fond par prednisolone/prednisone, avec ou sans traitement immunosuppresseur. Les doses de corticostéroïdes par voie orale ont été ajustées selon l'appréciation de l'investigateur.
Rémission
Les co-critères d'évaluation principaux étaient : la durée totale cumulée de rémission, définie par un BVAS (Birmingham Vasculitis Activity Score) = 0 et une dose de prednisolone/prednisone ≤ 4 mg/jour ; et la proportion de patients en rémission aux semaines 36 et 48 de la période de traitement de l'étude. Un BVAS=0 correspond à l'absence de vascularite active.
Par rapport au groupe placebo, une augmentation significative de la durée cumulée en rémission a pu être observée chez les patients recevant 300 mg de mépolizumab. De plus, une proportion significativement plus grande de patients recevant 300 mg de mépolizumab a été en rémission aux semaines 36 et 48 par rapport au groupe placebo (tableau 6).
Pour les deux co-critères d'évaluation principaux, l'effet bénéfique du traitement par 300 mg de mépolizumab a été observé par rapport au placebo, que les patients aient reçu ou non un traitement immunosuppresseur en plus de la corticothérapie de fond.
En utilisant comme définition de la rémission pour le critère d'évaluation secondaire (BVAS=0 et prednisolone/prednisone ≤ 7,5 mg/jour), la durée cumulée de la rémission était également significativement plus longue (p<0,001) et la proportion de patients en rémission aux semaines 36 et 48 plus large (p<0,001), pour les patients qui recevaient 300 mg de mépolizumab, par rapport au groupe placebo.
Tableau 6 : Analyse des co-critères d'évaluation principaux
| | Nombre (%) de patients | |
| Placebo N=68 | Mepolizumab (300 mg) N=68 | |
| Durée cumulée en rémission sur 52 semaines | | |
| 0 | 55 (81) | 32 (47) |
| >0 à <12 semaines | 8 (12) | 8 (12) |
| 12 à <24 semaines | 3 (4) | 9 (13) |
| 24 à <36 semaines | 0 | 10 (15) |
| ≥36 semaines | 2 (3) | 9 (13) |
| Odds ratio (mépolizumab/placebo) | | 5,91 |
| IC 95 % | --- | 2,68 ; 13,03 |
| Valeur du p | --- | <0,001 |
| Patients en rémission aux Semaines 36 et 48 | 2 (3) | 22 (32) |
| Odds ratio (mépolizumab/placebo) | | 16,74 |
| IC 95 % | --- | 3,61 ; 77,56 |
| Valeur du p | --- | <0,001 |
Un odds ratio >1 est en faveur du mépolizumab. Rémission : BVAS=0 et dose de corticothérapie orale ≤ 4mg / jour.
Rechute
Par rapport au placebo, le délai de survenue de la première rechute était significativement plus long chez les patients recevant 300 mg de mépolizumab (p<0,001). De plus, une réduction de 50 % du taux de rechutes annuel a été observée par rapport au placebo : respectivement 1,14 vs 2,27.
Réduction des corticostéroïdes oraux
Au cours des semaines 48-52, une diminution significative de la dose quotidienne moyenne de corticostéroïdes oraux a été observée chez les sujets traités par mépolizumab par rapport à ceux recevant le placebo. De la semaine 48 à la semaine 52, 59 % et 44 % des patients traités par mépolizumab ont réussi à réduire leur dose quotidienne moyenne de CSO respectivement ≤ 7,5 mg et ≤ 4 mg, comparé à 33 % et 7 % des patients du groupe placebo. 18 % des patients du groupe mépolizumab ont réussi à arrêter complètement leur corticothérapie orale par rapport à 3 % dans le groupe placebo.
Questionnaire de Contrôle de l'Asthme (Asthma Control Questionnaire) - 6 (ACQ-6)
Pendant les semaines 49 à 52, une amélioration significative de la moyenne du score ACQ a été rapportée chez les patients traités par mépolizumab par rapport aux patients recevant le placebo.
Syndrome hyperéosinophilique
L'étude 200622 était une étude de 32 semaines, randomisée, en double aveugle, contrôlée contre placebo qui a inclus 108 patients ≥ 12 ans présentant un syndrome hyperéosinophilique. Les patients recevaient soit 300 mg de mépolizumab soit du placebo, administré par voie sous-cutanée une fois toutes les 4 semaines concomitamment au traitement habituel de leur syndrome hyperéosinophilique. Dans l'étude 200622, les traitements du syndrome hyperéosinophilique incluaient entre autres : les corticostéroïdes oraux (CSO), les immunosuppresseurs, les traitements cytotoxiques ou d'autres traitements symptomatiques associés au syndrome hyperéosinophilique comme l'oméprazole.
Les patients entrant dans l'étude avaient connu au moins deux poussées de syndrome hyperéosinophilique dans les 12 derniers mois et avaient un taux d'éosinophiles sanguins ≥ 1000 cellules/µL pendant la sélection. Les patients présentant un réarrangement FIP1L1-PDGFRα étaient exclus de l'étude.
Le critère d'évaluation principal de l'étude 200622 était la proportion de patients ayant eu une poussée de syndrome hyperéosinophilique au cours des 32 semaines de traitement. Une poussée de syndrome hyperéosinophilique est définie comme une aggravation des signes cliniques et des symptômes de syndrome hyperéosinophilique nécessitant l'augmentation de la posologie des CSO ou l'augmentation de la posologie/ajout d'un traitement cytotoxique ou d'immunosuppresseur utilisés dans le traitement du syndrome hyperéosinophilique, ou bien nécessitant une dose de CSO en aveugle en raison d'une augmentation des éosinophiles sanguins (à 2 occasions ou plus).
L'analyse principale a comparé les patients qui ont présenté une poussée de syndrome hyperéosinophilique ou qui ont arrêté l'étude, dans les groupes mépolizumab et placebo. Sur les 32 semaines de traitement, 50% de patients en moins ont présenté une poussée de syndrome hyperéosinophilique ou ont arrêté l'étude lorsqu'ils étaient traités avec 300 mg de mépolizumab par rapport au placebo ; 28% versus 56% respectivement (OR 0,28, IC 95% : 0,12 ;0,64) (voir tableau 8). Les critères d'évaluation secondaires étaient : le délai de survenue de la première poussée du syndrome hyperéosinophilique, la proportion de patients ayant eu une poussée de syndrome hyperéosinophilique au cours des semaines 20 à 32, le taux de poussées de syndrome hyperéosinophilique et la variation par rapport à l'inclusion de la gravité de la fatigue.
Tous les critères d'évaluation secondaires étaient statistiquement significatifs et confortaient le critère principal (voir Figure 2 et Tableau 8).
Tableau 7 : Résultats du critère principal/analyse dans la population en Intention de Traiter (Etude 200622)
| | Mépolizumab 300 mg N= 54 | Placebo N= 54 |
| Proportion de patients ayant eu une poussée de syndrome hyperéosinophilique | ||
| Patients avec ≥1 poussée de syndrome hyperéosinophilique ou qui ont arrêté l'étude (%) | 15 (28) | 30 (56) |
| | Mépolizumab 300 mg N= 54 | Placebo N= 54 |
| Patients avec ≥1 poussée de syndrome hyperéosinophilique (%) | 14 (26) | 28 (52) |
| Patients sans poussée de syndrome hyperéosinophilique qui ont arrêté l'étude (%) | 1 (2) | 2 (4) |
| Odds ratio (IC 95%) | 0.28 (0.12, 0.64) | |
| CMH p-value | 0.002 | |
CMH : Cochran-Mantel-Haenszel
Délai de survenue de la première poussée
Les patients qui ont reçu 300 mg de mépolizumab ont présenté une augmentation significative du délai de survenue de la première poussée de syndrome hyperéosinophilique comparé à ceux qui ont reçu le placebo. Le risque de première poussée de syndrome hyperéosinophilique pendant la période de traitement était 66% inférieur pour les patients traités avec Nucala par rapport à ceux recevant le placebo (Hazard Ratio : 0,34 ; IC 95 % 0,18 ; 0.67; p=0,002).
Figure 2 : Courbe de Kaplan Meier pour le délai de survenue de la première poussée de syndrome hyperéosinophilique
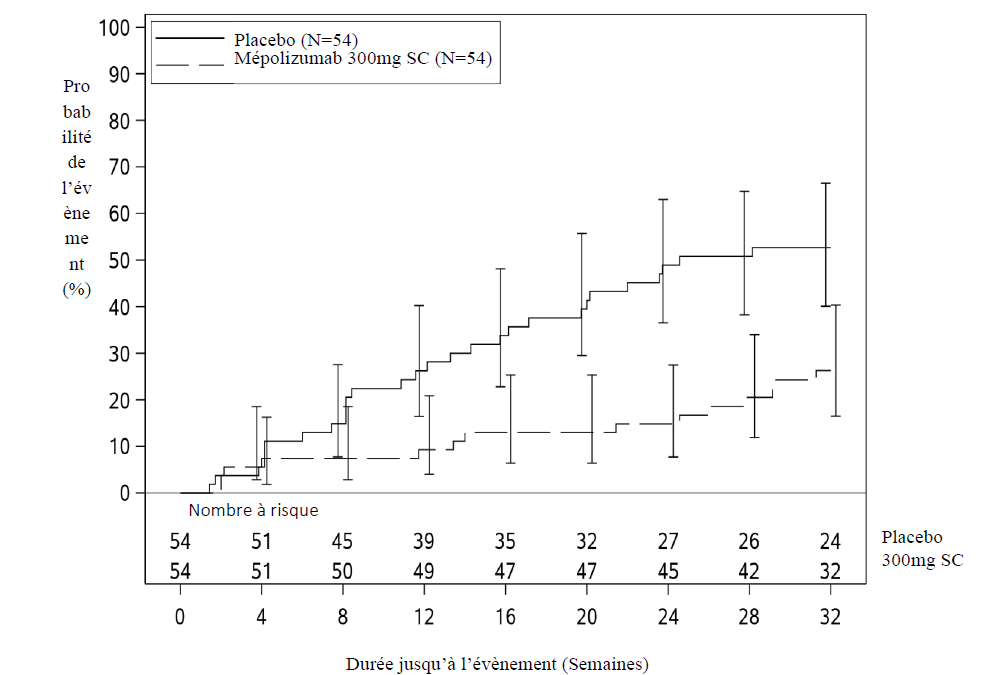
Tableau 8 : Résultats des autres critères secondaires dans la population en intention de traiter (Etude 200622)
| | Mépolizumab 300 mg N= 54 | Placebo N= 54 |
| Poussées de syndrome hyperéosinophilique au cours des semaines 20 à 32 | ||
| Patients avec ≥1 poussée de syndrome hyperéosinophilique ou qui ont arrêté l'étude (%) | 9 (17) | 19 (35) |
| Odds ratio (IC 95%) | 0,33 (0,13;0,85) | |
| CMH p-value | 0,02 | |
| Taux de poussées de syndrome hyperéosinophilique | ||
| Taux moyen estimé/an | 0,50 | 1,46 |
| Ratio des taux (IC 95%)a | 0,34 (0,19; 0,63) | |
| Test de la somme des rangs de Wilcoxon Valeur de p | 0,002 | |
| Variation entre l'inclusion et la semaine 32 de la gravité de la fatigue basée sur l'item 3 du Brief Fatigue Inventory (BFI) (plus mauvais niveau de fatigue au cours des dernières 24h)b | ||
| Variation médiane de l'item 3 du BFI | -0,66 | 0,32 |
| Comparaison (mepolizumab vs. placebo) Test de la somme des rangs de Wilcoxon Valeur de p | 0,036 | |
a Ratio des taux <1 en faveur de mépolizumab.
b les patients avec des données manquantes incluaient la pire valeur observée. Item 3 du BFI : 0 = aucune fatigue à 10 = plus mauvais niveau de fatigue imaginable
CMH = Cochran-Mantel-Haenszel
Etude d'extension en ouvert
L'étude 205203 de 20 semaines était une étude d'extension en ouvert de l'étude 200622. Il était permis d'ajuster le traitement du syndrome hyperéosinophilique selon les traitements standards locaux tout en maintenant le traitement par mépolizumab à 300 mg en commençant à la semaine 4. Dans cette étude, l'effet du traitement par mépolizumab sur la réduction des poussées de syndrome hyperéosinophilique rapportées pendant l'étude 200622 était maintenu pour les patients qui avaient continué le traitement par mépolizumab dans l'étude 205203, parmi ceux-ci, 94% (47/50) des patients n'ont pas présenté de poussée.
Parmi les 72 patients nécessitant un traitement par CSO pendant les semaines 0 à 4 de l'étude d'extension, 28% des patients ont atteint une réduction de la dose de CSO quotidienne moyenne de ≥ 50% pendant les semaines 16 à 20.
Population pédiatrique
Asthme sévère réfractaire à éosinophiles
34 adolescents (âgés de 12 ans à 17 ans) ont été inclus dans l'étude MEA115588 et dans l'étude en double-aveuble contrôlée contre placebo 200862. Sur ces 34 patients, 12 ont reçu le placebo, 9 ont reçu 75 mg de mépolizumab par voie intraveineuse et 13 ont reçu 100 mg de mépolizumab par voie sous-cutanée. Une analyse combinée de ces études a montré une réduction cliniquement significative de 40 % des exacerbations chez les adolescents traités par mépolizumab par rapport à ceux recevant le placebo (ratio 0,60 ; IC 95 % : 0,17 ; 2,10).
Granulomatose éosinophilique avec polyangéite
Il n'y a pas de données cliniques disponibles chez les enfants et les adolescents âgés de 6 à 17 ans.
Syndrome hyperéosinophilique
4 adolescents (âgés de 12 à 17 ans) ont été inclus dans l'étude 200622 ; 1 adolescent a reçu du mépolizumab 300 mg et 3 adolescents ont reçu du placebo pendant 32 semaines. L'adolescent traité avec mépolizumab dans l'étude 200622 de 32 semaines n'a pas eu de poussée de syndrome hyperéosinophilique. Les 4 adolescents qui ont terminé l'étude 200622 ont continué dans l'étude d'extension en ouvert de 20 semaines 205203 au cours de laquelle 1 adolescent sur les 4 a eu une poussée de syndrome hyperéosinophilique.
La pharmacocinétique du mépolizumab administré par voie sous-cutanée à des patients présentant un asthme et une polypose naso-sinusienne apparaît linéaire pour des doses comprises entre 12,5 mg et 250 mg.
L'administration de 300 mg de mépolizumab par voie sous-cutanée a permis d'obtenir une exposition systémique d'environ trois fois celle obtenue avec 100 mg de mépolizumab.
Absorption
Après administration sous-cutanée à des sujets sains ou à des patients asthmatiques, le mépolizumab a été absorbé lentement, avec un temps médian pour atteindre la concentration plasmatique maximale (Tmax) compris entre 4 et 8 jours.
Après une injection unique par voie sous-cutanée soit au niveau de l'abdomen, soit au niveau de la cuisse ou du bras de sujets sains, la biodisponibilité absolue du mépolizumab a été respectivement de 64 %, 71 % et 75 %. Chez les patients asthmatiques, la biodisponibilité absolue du mépolizumab administré par voie sous-cutanée au niveau du bras a été comprise entre 74 % et 80 %.
L'administration sous-cutanée de doses répétée toutes les 4 semaines entraîne une accumulation de l'exposition environ deux fois plus élevée à l'état d'équilibre.
Distribution
Après une injection unique par voie intraveineuse chez des patients asthmatiques, le mépolizumab se répartit dans un volume moyen de distribution compris entre 55 et 85 ml/kg.
Biotransformation
Le mépolizumab est un anticorps monoclonal IgG1 humanisé. Il est dégradé par des enzymes protéolytiques réparties dans tout l'organisme et ne se limitant pas au tissu hépatique.
Élimination
Après une injection unique par voie intraveineuse chez des patients asthmatiques, la clairance (Cl) systémique moyenne a été comprise entre 1,9 et 3,3 ml/jour/kg, avec une demi-vie terminale moyenne (t1/2) d'environ 20 jours. La demi-vie terminale moyenne après une injection sous-cutanée de mépolizumab a été comprise entre 16 et 22 jours. La clairance systémique estimée du mépolizumab dans l'analyse de pharmacocinétique de population a été de 3,1 ml/jour/kg.
Populations spécifiques
Patients âgés (≥ 65 ans)
Les données pharmacocinétiques disponibles chez les patients âgés (≥ 65 ans) dans l'ensemble des études cliniques sont limitées (n=90). Cependant, l'analyse de pharmacocinétique de population n'a mis en évidence aucun effet de l'âge sur la pharmacocinétique du mépolizumab dans la tranche d'âge 12-82 ans.
Insuffisance rénale
Aucune étude spécifique visant à évaluer l'effet d'une insuffisance rénale sur la pharmacocinétique du mépolizumab n'a été conduite. Au vu des analyses de pharmacocinétique de population, aucune adaptation posologique n'apparait nécessaire chez les patients présentant une clairance de la créatinine comprise entre 50 et 80 ml/min. Les données disponibles sont limitées pour les patients présentant une clairance de la créatinine < 50 ml/min.
Insuffisance hépatique
Aucune étude spécifique visant à évaluer l'effet d'une insuffisance hépatique sur la pharmacocinétique du mépolizumab n'a été conduite. Le mépolizumab étant dégradé par des enzymes protéolytiques réparties dans tout l'organisme et ne se limitant pas au tissu hépatique, il est peu probable que des modifications de la fonction hépatique aient un retentissement sur l'élimination du mépolizumab.
Population pédiatrique
Asthme sévère à éosinophiles et syndrome hyperéosinophilique
Les données pharmacocinétiques disponibles dans la population pédiatrique sont limitées (59 patients ayant une œsophagite à éosinophiles, 55 patients ayant un asthme sévère réfractaire à éosinophiles et 1 patient ayant un syndrome hyperéosinophilique). Les paramètres pharmacocinétiques du mépolizumab administré par voie intraveineuse ont été évalués au cours d'une analyse de pharmacocinétique de population chez des patients âgés de 2 à 17 ans présentant une œsophagite à éosinophiles. Les paramètres pharmacocinétiques chez l'enfant ont été ceux attendus par extrapolation à partir de ceux mesurés chez l'adulte après ajustement en fonction du poids corporel. Les paramètres pharmacocinétiques du mépolizumab chez les adolescents ayant un asthme sévère réfractaire à éosinophiles ou un syndrome hyperéosinophilique, inclus dans les études de phase III, avaient le même profil que celui observé chez l'adulte (voir rubrique Posologie et mode d'administration).
La pharmacocinétique du mépolizumab administré par voie sous-cutanée a été étudiée chez des patients pédiatriques âgés de 6 ans à 11 ans, présentant un asthme sévère réfractaire à éosinophiles, dans une étude en ouvert et non contrôlée de 12 semaines. Les données de pharmacocinétique chez les patients pédiatriques concordaient avec celles observées chez l'adulte et l'adolescent, après ajustement sur le poids et la biodisponibilité. Chez 76 % des patients étudiés, la biodisponibilité sous- cutanée absolue semble être identique à celle observée chez l'adulte et l'adolescent. L'exposition systémique après administration sous-cutanée de soit 40 mg (pour un poids < 40 kg), soit 100 mg (pour un poids ≥ 40 kg) était respectivement augmentée d'un facteur 1,32 et 1,97 par rapport à celle observée chez l'adulte avec la dose de 100 mg.
L'étude par modélisation pharmacocinétique et par simulation d'une dose de 40 mg de mépolizumab administrée par voie sous-cutanée toutes les 4 semaines chez des enfants de 6 ans à 11 ans, dans une tranche de poids corporel de 15 à 70 kg suggère que l'exposition systémique obtenue avec ce schéma
posologique se maintiendrait en moyenne dans les 38 % de celle observée chez l'adulte avec une dose de 100 mg. Ce schéma posologique a été considéré comme acceptable compte-tenu du large index thérapeutique du mépolizumab.
Granulomatose éosinophilique avec polyangéite
Les données de pharmacocinétique du mépolizumab chez les enfants (âgés de 6 à 17 ans) atteints de granulomatose éosinophilique avec polyangéite ont été établies par modélisation et simulation, sur la base des données de pharmacocinétique recueillies dans d'autres pathologies Il est attendu qu'elles soient similaires à celles observées chez les enfants présentant un asthme sévère à éosinophiles. La posologie recommandée chez l'enfant âgé de 6 à 11 ans et pesant entre 15 et 70 kg permet d'anticiper une exposition en moyenne équivalente à 26 % de celle d'un adulte recevant une dose de 300 mg.
Nucala n'a aucun effet ou a un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Le mépolizumab étant un anticorps monoclonal, aucune étude de génotoxicité ou de carcinogénicité n'a été menée.
Toxicologie et/ou pharmacologie chez l'animal
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité ou de toxicologie après administration répétée chez le singe, n'ont pas révélé de risque particulier pour l'homme. L'administration intraveineuse et sous-cutanée à des singes a été associée à une réduction des taux d'éosinophiles dans le sang périphérique et les poumons, sans effet toxicologique.
Les éosinophiles sont supposés être impliqués dans la réponse immunitaire à certaines parasitoses. Des études menées chez des souris traitées par des anticorps anti-IL-5 ou génétiquement déficientes en IL-5 ou en éosinophiles n'ont montré aucune difficulté à lutter contre des parasitoses. La pertinence de ces observations pour l'homme n'est pas connue.
Fertilité
Aucun trouble de la fertilité n'a été observé dans l'étude de toxicité sur la fertilité et sur la reproduction générale menée chez la souris avec un anticorps analogue inhibant l'IL-5 de la souris. Cette étude n'incluait pas d'évaluation sur la portée ni d'évaluation fonctionnelle sur la progéniture.
Grossesse
Chez le singe, le mépolizumab n'a eu aucun impact sur la grossesse ou sur le développement embryonnaire/fœtal et postnatal (y compris sur la fonction immunitaire) de la progéniture. Aucune recherche de malformations internes ou squelettiques n'a été réalisée. Les données obtenues chez le singe Cynomolgus montrent que le mépolizumab a traversé le placenta. Les concentrations en mépolizumab observées dans la progéniture étaient environ 1,2 à 2,4 fois supérieures à celles observées chez les mères pendant plusieurs mois après la mise bas, sans impact délétère sur le système immunitaire de la progéniture.
La reconstitution doit être réalisée en respectant les règles d'asepsie.
Instructions concernant la reconstitution de chaque flacon
Reconstituer le contenu du flacon avec 1,2 ml d'eau stérile pour préparations injectables en utilisant de préférence une seringue graduée de 2 à 3 ml et une aiguille de calibre 21 gauges. Le jet d'eau stérile doit être orienté à la verticale au centre du lyophilisat. Garder le flacon à température ambiante pendant la reconstitution, remuer doucement le flacon en réalisant un mouvement circulaire pendant 10 secondes toutes les 15 secondes jusqu'à dissolution de la poudre.
Remarque : la solution reconstituée ne doit pas être secouée pendant la procédure de reconstitution, car cela risque d'entraîner la formation de mousse ou la précipitation du produit. La reconstitution est habituellement terminée dans les 5 minutes qui suivent l'ajout d'eau stérile, mais un temps plus long peut être nécessaire.
Si un dispositif mécanique (agitateur) est utilisé pour reconstituer Nucala, la reconstitution peut être obtenue avec une vitesse de rotation de 450 tours/min pendant une durée maximale de 10 minutes. Il est également possible d'appliquer une vitesse de rotation de 1 000 tours/min pendant une durée maximale de 5 minutes.
Après la reconstitution, une inspection visuelle de Nucala doit être réalisée afin de chercher la présence de particules et de vérifier la limpidité de la solution avant utilisation. La solution reconstituée doit être transparente à opalescente et incolore à jaune pâle ou brun pâle, sans particules visibles. De petites bulles d'air peuvent cependant se former et sont acceptables. Une solution contenant des particules ou ayant un aspect trouble ou laiteux ne doit pas être utilisée.
Si la solution reconstituée n'est pas utilisée immédiatement, elle doit être :
Conservée à l'abri de la lumière
Conservée à une température ne dépassant pas 30°C, ne pas être congelée
Eliminée si elle n'est pas utilisée dans les 8 heures qui suivent la reconstitution
Instructions concernant l'administration d'une dose de 100 mg
Pour l'injection sous-cutanée, il est recommandé d'utiliser une seringue graduée de 1 ml en polypropylène munie d'une aiguille à usage unique de 13 mm (0,5 pouce) et de calibre 21 à 27 gauges.
Juste avant l'administration, prélever 1 ml de la solution reconstituée de Nucala. Ne pas secouer la solution reconstituée pendant la procédure de reconstitution, car cela pourrait entraîner la formation de mousse ou la précipitation du produit.
Administrer 1 ml de solution injectable (équivalent à 100 mg de mépolizumab) par voie sous- cutanée soit dans la partie supérieure du bras, soit au niveau de la cuisse ou de l'abdomen.
S'il est nécessaire d'utiliser plus d'un flacon pour l'administration de la dose prescrite, répéter les étapes 1 à 3. Il est recommandé que les sites d'injection soient espacés d'au moins 5 cm.
Instructions pour l'administration d'une dose de 40 mg
Pour l'injection sous-cutanée, il est recommandé d'utiliser une seringue graduée de 1 ml en polypropylène munie d'une aiguille à usage unique de 13 mm (0,5 pouce) et de calibre 21 à 27 gauges.
Juste avant l'administration, prélever 0,4 ml de la solution reconstituée de Nucala. Ne pas secouer la solution reconstituée pendant la procédure de reconstitution car cela pourrait entraîner la formation de mousse ou la précipitation du produit. Jeter le reste de la solution.
Administrer 0,4 mL de solution injectable (équivalent à 40 mg de mépolizumab) par voie sous- cutanée soit dans la partie supérieure du bras, soit au niveau de la cuisse ou de l'abdomen.
Élimination
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
liste I
prescription initiale réservée à certains spécialistes
renouvellement de la prescription réservé aux spécialistes en ALLERGOLOGIE, DERMATOLOGIE, HEMATOLOGIE, MEDECINE INTERNE, OTO-RHINO-LARYNGOLOGIE, PNEUMOLOGIE et PEDIATRIE.
Médicament d'exception.
Remboursement en fonction de l'indication (JO du 28/10/2022) :
Les seules indications thérapeutiques ouvrant droit à la prise en charge ou au remboursement par l'assurance maladie sont :
. Asthme sévère à éosinophiles chez l'adulte:
NUCALA est indiqué en traitement additionnel dans l'asthme sévère réfractaire à éosinophiles chez les adultes, répondant aux critères suivants:
- un taux d'éosinophiles sanguins ≥ 150/µL dans les douze derniers mois; ET
- au moins deux épisodes d'exacerbations asthmatiques ayant nécessité un traitement par corticoïde oral (≥ 3 jours chacun) dans les 12 derniers mois malgré un traitement de fond associant des corticoïdes inhalés à dose élevée et un bronchodilatateur d'action longue (LABA);
- ou un traitement par corticothérapie orale pendant au moins 6 mois au cours des 12 derniers mois. Les patients dont l'asthme n'est pas contrôlé en raison d'un traitement de fond inadapté, de problèmes d'observance, de comorbidités ou de facteurs de risque aggravants non pris en charge n'entrent pas dans ce périmètre.
. Asthme sévère à éosinophiles chez l'adolescent et l'enfant de 6 ans et plus:
NUCALA est indiqué en
traitement additionnel dans l'asthme sévère réfractaire à éosinophiles
chez les adolescents et les enfants de 6 ans et plus répondant aux
critères suivants:
- un taux d'éosinophiles sanguins ≥ 300/µL dans les douze derniers mois; ET
- au moins deux
épisodes d'exacerbations asthmatiques ayant nécessité un traitement par
corticoïde oral (≥ 3 jours chacun) dans les 12 derniers mois malgré un
traitement de fond associant des corticoïdes inhalés à dose élevée et
un bronchodilatateur d'action longue (LABA);
- ou un traitement par
corticothérapie orale pendant au moins 6 mois au cours des 12 derniers
mois. Les patients dont l'asthme n'est pas contrôlé en raison d'un
traitement de fond inadapté, de problèmes d'observance, de comorbidités
ou de facteurs de risque aggravants non pris en charge n'entrent pas
dans ce périmètre.
. Polypose
naso-sinusienne:
NUCALA est indiqué en traitement additionnel aux
corticostéroïdes par voie nasale chez les patients adultes présentant
une polypose naso-sinusienne sévère insuffisamment contrôlés par des
corticostéroïdes systémiques et la chirurgie.
. Granulomatose
éosinophilique avec polyangéite:
NUCALA est indiqué chez les patients
âgés de 6 ans et plus, en traitement additionnel des formes
récidivantes ou réfractaires de la granulomatose éosinophilique avec
polyangéite.
. Syndrome
hyperéosinophilique:
NUCALA est indiqué, en traitement additionnel,
chez les patients adultes qui présentent un syndrome
hyperéosinophilique lymphoïde ou idiopathique insuffisamment contrôlé.
Poudre pour solution injectable. Poudre blanche lyophilisée.
Flacon de 10 ml en verre transparent incolore de type I, muni d'un bouchon en caoutchouc bromobutyl serti d'une bague en aluminium gris dotée d'un opercule en plastique, contenant 100 mg de poudre pour solution injectable.
Présentation :
1 flacon.
Chaque flacon contient 100 mg de mépolizumab. Après reconstitution, chaque ml de solution contient 100 mg de mépolizumab.
Le mépolizumab est un anticorps monoclonal humanisé produit par la technologie de l'ADN recombinant dans des cellules ovariennes de hamsters chinois.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Saccharose
Phosphate de sodium dibasique heptahydraté
Polysorbate 80