
JETREA 0,5 mg-0,2 ml, solution à diluer injectable, emballage de 1 flacon de 0,20 mL
Retiré du marché le : 15/12/2020
Dernière révision : 08/01/2019
Taux de TVA : 2.1%
Laboratoire exploitant : ALCON
Source :

JETREA est indiqué chez les adultes pour le traitement de la traction vitréo-maculaire (TVM), notamment lorsqu'elle est associée à un trou maculaire d'un diamètre inférieur ou égal à 400 microns (voir rubrique Propriétés pharmacodynamiques).
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Composition.
Infections oculaires ou périoculaires actives ou suspectées.
Suivi après injection
JETREA est administré par injection intravitréenne uniquement. Les injections intravitréennes peuvent être associées à une inflammation/infection intraoculaire, une hémorragie intraoculaire et à une augmentation de la pression intraoculaire (PIO). Des techniques d'injection aseptiques adéquates doivent toujours être utilisées. Suite à l'injection intravitréenne, les patients doivent être surveillés pour tout effet indésirable, tel que (entre autres) l'inflammation/infection intraoculaire et l'augmentation de la PIO. Des augmentations transitoires de la PIO, notamment une cécité transitoire et la non-perfusion du nerf optique, ont été observées dans les 60 minutes suivant l'injection de JETREA. Le suivi des augmentations de la PIO peut inclure une vérification de la perfusion de la tête du nerf optique immédiatement après l'injection et une tonométrie dans les 30 minutes suivant l'injection. L'inflammation/infection intraoculaire peut être évaluée à l'aide d'une biomicroscopie pratiquée entre deux et sept jours après l'injection. Les patients doivent être informés que tout symptôme évocateur d'une inflammation/infection ou tout autre symptôme visuel ou oculaire doit être signalé sans délai. Si l'un des événements ci-dessus se produit, le patient doit être traité conformément aux pratiques médicales standard en vigueur.
Traitement bilatéral
La sécurité et l'efficacité de JETREA administré simultanément dans les deux yeux n'ont pas été étudiées. Par conséquent, un traitement bilatéral de façon simultanée n'est pas recommandé.
Administration répétée
L'administration répétée de JETREA dans le même œil n'a pas été étudiée de manière adéquate et elle n'est donc pas recommandée.
Population avec peu ou pas de données
JETREA n'a pas été étudié chez les patients présentant des trous maculaires de grand diamètre (> 400 microns), une myopie élevée (correction sphérique > 8 dioptries ou longueur axiale > 28 mm), une aphakie, un antécédent de décollement reghmatogène de la rétine, une instabilité zonulaire du cristallin, une chirurgie oculaire ou une injection intraoculaire récentes (notamment un traitement par laser), une rétinopathie diabétique proliférante, des rétinopathies ischémiques, des occlusions veineuses rétiniennes, une dégénérescence maculaire exsudative liée à l'âge (DMLA) et une hémorragie vitréenne. Chez ces patients, le traitement n'est pas recommandé.
L'expérience chez les patients présentant une rétinopathie diabétique non proliférante ou des antécédents d'uvéite (y compris une inflammation active sévère) ou de traumatisme oculaire important est limitée. Il convient d'être prudent lors d'un traitement chez ces patients.
Autre
La possibilité d'une subluxation du cristallin ou de phacodonésis ne peut pas être exclue. Si cet évènement se produit, il doit être traité conformément aux pratiques médicales standard en vigueur. Les patients doivent être surveillés de manière appropriée (voir rubriques Effets indésirables et Données de sécurité précliniques).
L'effet de l'ocriplasmine (en particulier dans la résolution de l'adhérence vitréo-maculaire ou en provoquant un décollement postérieur du vitré total [DPV]) est réduit chez les sujets présentant une membrane épirétinienne (MER) ou un diamètre de l'AVM > 1500 microns (voir rubrique Propriétés pharmacodynamiques).
Il existe un risque de diminution importante de l'acuité visuelle durant la première semaine suivant l'injection. Les patients doivent être surveillés de manière appropriée (voir rubrique Effets indésirables).
Les examens ophtalmologiques peuvent être anormaux après l'administration de JETREA. Ceux-ci comprennent la tomographie par cohérence optique (OCT), l'ophtalmoscopie (reflet fovéal), le test de vision des couleurs (test 28 Hue de Roth) et l'ERG plein champ. Cela doit être pris en compte lorsque ces tests sont mis en œuvre pour le diagnostic ou la surveillance d'autres pathologies (voir
rubrique Effets indésirables).
Résumé du profil de sécurité
Plus de 1400 patients ont été traités avec la dose recommandée de 0,125 mg de JETREA dans des études cliniques interventionnelles.
Tous les effets indésirables étaient oculaires. Dans 3 études cliniques comportant un suivi allant de 6 mois (TG-MV-006 et TG-MV-007) à 24 mois (TG-MV-014), les effets indésirables les plus fréquemment signalés étaient les corps flottants vitréens, la douleur oculaire, la photopsie et la chromatopsie, ainsi que l'hémorragie conjonctivale résultant de la procédure d'injection. La plupart des effets indésirables se sont produits la première semaine après l'injection. La majorité de ces effets étaient non graves, d'intensité légère à modérée et se sont résolus dans les deux à trois semaines.
Les informations relatives à la résolution d'événements spécifiques, tels que la chromatopsie et les modifications de l'ERG, sont fournies dans le paragraphe correspondant de la rubrique « Description de certains effets indésirables ».
Les effets indésirables les plus importants sur le plan clinique incluaient la cécité transitoire, la déchirure rétinienne, le décollement de la rétine, la subluxation du cristallin et l'aggravation du trou maculaire.
Tableau des effets indésirables
Le tableau suivant récapitule les effets indésirables rapportés dans l'œil traité durant les études cliniques et/ou rapportés depuis la commercialisation.
Des symptômes visuels perçus dans l'œil controlatéral ou de manière bilatérale ont été également signalés.
Les effets indésirables avec une possibilité réelle de lien de causalité avec la procédure d'injection ou JETREA sont listés ci-dessous selon la terminologie MedDRA par classe de systèmes d'organes et par fréquence, selon les règles suivantes: très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Parmi chaque groupe de fréquence, les effets indésirables sont classés par ordre de gravité décroissante.
| Affections oculaires | Très fréquent Corps flottants vitréens, douleur oculaire, hémorragie conjonctivale, chromatopsie* Fréquent Acuité visuelle réduite*, altération visuelle1), anomalie du champ visuel2), vision trouble, hémorragie rétinienne, hémorragie vitréenne, trou maculaire*, dégénérescence maculaire, dégénérescence rétinienne, œdème maculaire3), œdème rétinien4), épithéliopathie pigmentaire rétinienne, métamorphopsie, œdème conjonctival, œdème de la paupière, inflammation du vitré, cellules dans la chambre antérieure, effet Tyndall dans la chambre antérieure, inflammation de l'iris, photopsie, hyperhémie conjonctivale, hyperhémie oculaire, décollement vitréen, irritation de l'œil, œil sec, sensation de corps étranger dans l'œil, prurit oculaire, gêne oculaire, photophobie, augmentation de la sécrétion lacrymale Peu fréquent Cécité transitoire, subluxation du cristallin*, déchirure rétinienne*5), décollement de la rétine*5), cécité nocturne, réflexe pupillaire affaibli, diplopie, hyphéma, myosis, pupilles inégales, abrasion de la cornée, inflammation de la chambre antérieure, inflammation oculaire, irritation conjonctivale |
| Investigations | Très fréquent Rétinogramme anormal*, test de vision des couleurs anormal† Fréquent Augmentation de la pression intraoculaire, reflet maculaire anormal, tomographie par cohérence optique (OCT) anormale* |
* voir rubrique « Description de certains effets indésirables ».
1) comprenant une vision obscurcie
2) comprenant un scotome
3) comprenant un œdème maculaire cystoïde
4) comprenant du liquide sous-rétinien
5) événements survenant avant la vitrectomie
† utilisant le test de vision des couleurs 28 Hue de Roth. Voir également rubrique Mises en garde et précautions d'emploi.
Description de certains effets indésirables
Acuité visuelle réduite
Dans les études pivot contrôlées de phase III contre placebo (TG-MV-006 et TG-MV-007), chez 7,7% des patients sous JETREA et 1,6% des patients recevant un placebo, une baisse aigüe de la meilleure acuité visuelle corrigée (MAVC) ≥ 2 lignes (≥ 10 lettres ETDRS) a été observée au cours de la première semaine suivant l'injection, sans autre explication à ce changement. La baisse de l'acuité visuelle s'était résolue à la fin des études pour la majorité des patients sous JETREA (80,6%), mais certains patients n'avaient pas récupéré malgré une vitrectomie. Le délai médian de résolution était de 22 jours.
Dans l'étude TG-MV-014, chez 2,8% des patients sous JETRA et 1,4% des patients sous injection simulée, une baisse aigüe de la MAVC ≥ 2 lignes a été observée au cours de la première semaine suivant l'injection. Sur les 4 patients sous JETREA présentant une baisse aigüe de l'acuité visuelle, 3 ont récupéré après la vitrectomie. Voir rubrique Mises en garde et précautions d'emploi pour les recommandations de surveillance.
Chromatopsie (dont dyschromatopsie et test de vision des couleurs anormal)
Des altérations de la vision des couleurs (comprenant une vision jaunâtre et un test de vision des couleurs 28 Hue de Roth anormal) ont été signalées comme un effet indésirable très fréquent chez des patients ayant reçu une injection de JETREA. La majorité des événements étaient non graves, d'intensité légère et se sont généralement résolus spontanément. Le délai médian de résolution était de trois mois.
Rétinogramme anormal
Des modifications des enregistrements électrorétinographiques (ERG) (diminution de l'amplitude des ondes a et b) ont été signalées comme un effet indésirable très fréquent chez des patients ayant reçu une injection de JETREA ; dans la majorité des cas, une altération visuelle et une chromatopsie ont également été rapportées.
Dans l'étude TG-MV-014, un sous-ensemble de 40 patients recevant JETREA ont systématiquement fait l'objet d'examens ERG ; les modifications de l'ERG qui étaient survenues chez 16 des 40 patients se sont résolues pour la majorité d'entre eux (13 patients sur 16). Le délai médian de résolution était de six mois. Les modifications de l'ERG n'étaient pas prédictives de résultats négatifs en termes d'acuité visuelle ; l'acuité visuelle s'est améliorée ou maintenue chez 15 patients sur 16 par rapport au début de l'étude.
Déchirures et décollement de la rétine
Dans les études pivot contrôlées de phase III contre placebo (TG-MV-006 et TG-MV-007), des déchirures et décollement de la rétine ont été signalés chez 1,9% des patients ayant reçu une injection de JETREA contre 4,3% ayant reçu une injection de placebo. Dans les deux groupes, la plupart de ces événements se sont produits pendant ou après la vitrectomie. L'incidence des décollements rétiniens s'étant produits avant la vitrectomie était de 0,4% dans le groupe de JETREA et nulle dans le groupe placebo.
Dans l'étude TG-MV-014, une déchirure rétinienne a été signalée chez 1,4% des patients ayant reçu une injection de JETREA et 6,8% des patients ayant reçu une injection simulée ; l'incidence d'un décollement rétinien était de 1,4% dans les deux bras. Dans le groupe injection simulée, aucun événement ne s'est produit avant la vitrectomie. Dans le groupe JETREA, 1 patient (0,7%) a présenté une déchirure rétinienne et un décollement de la rétine entre le jour 0 et le jour 7 suivant l'injection.
Trou maculaire
Dans les études pivot contrôlées de phase III contre placebo (TG-MV-006 et TG-MV-007), des événements de trou maculaire (incluant à la fois des cas de progression et d'apparition) ont été signalés pour 6,7 % des patients ayant reçu une injection de JETREA contre 9,6 % des patients ayant reçu une injection de placebo au mois 6.
Dans l'étude TG-MV-014, des événements de trou maculaire (incluant à la fois des cas de progression et d'apparition) ont été signalés chez 15,8% des patients ayant reçu une injection de JETREA contre 13,5% des patients ayant reçu une injection simulée au mois 24.
Les taux de progression précoce de trou maculaire de pleine épaisseur (jusqu'au jour 7 après l'injection) au niveau de l'EPR (épithélium pigmentaire de la rétine) étaient plus élevés chez les patients traités par JETREA que chez les patients ayant reçu une injection simulée ou un placebo. Cependant, les taux de progression après le mois 6 étaient plus élevés chez les patients ayant reçu une injection simulée ou un placebo que chez les patients traités par JETREA. Toute persistance ou progression de trou maculaire doit être traitée conformément aux pratiques habituelles.
Subluxation du cristallin/phacodonésis
Un cas de subluxation du cristallin/phacodonésis a été signalé dans des études cliniques chez des adultes et semble être probablement lié au traitement de JETREA. Dans une étude clinique pédiatrique évaluant JETREA comme adjuvant à la vitrectomie, un cas de subluxation a été signalé chez un nourrisson prématuré qui avait reçu une injection intravitréenne unique de JETREA à 0,175 mg. Une subluxation du cristallin a été observée chez trois espèces animales à des concentrations d'ocriplasmine supérieures à la concentration clinique prévue dans l'AMM (voir rubrique Données de sécurité précliniques).
Selon l'activité protéolytique de l'ocriplasmine, les résultats précliniques et cliniques, le potentiel de subluxation du cristallin ou de phacodonésis ne peut être exclu. Si cet événement se produit, il doit être traité conformément aux pratiques médicales standard.
Tomographie par cohérence optique anormale
Dans l'étude TG-MV-014, une couche Segment Interne/Segment Externe (IS/OS), également appelée
« zone ellipsoïde », incomplète dans la zone centrale était très fréquente au début de l'étude (65,8% dans le groupe JETREA et 62,2% dans le groupe injection simulée). Toutefois, après le traitement, une proportion plus élevée de patients du groupe JETREA est passée d'une couche IS/OS intacte au début
de l'étude à une couche IS/OS incomplète dans la zone centrale par la suite, comparée au groupe injection simulée (7,7% et 2,8%, respectivement, au jour 28). Au-delà de la zone centrale, des aspects anormaux de la couche IS/OS attribués à JETREA ont été observés chez près de 10% des patients.
Des ruptures de la zone ellipsoïde à l'intérieur et à l'extérieur de la zone centrale ont été signalées dans des études non interventionnelles et des rapports d'évaluation post-commercialisation. Dans la majorité des cas, le problème s'est résolu en 6 mois. Du liquide sous-rétinien ainsi que des signes et symptômes d'altération de la fonction photoréceptrice, dont une diminution de l'acuité visuelle (dans certains cas sévères), ont été signalés en association avec ces événements.
Voir rubrique Mises en garde et précautions d'emploi pour les recommandations relatives au suivi. Une surveillance de routine est recommandée dans toutes les situations citées ci-dessus.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
SURVEILLANCE des effets indésirables, tel que l'inflammation/infection intraoculaire (biomicroscopie pratiquée entre deux et sept jours après l'injection) et l'augmentation de la PIO (vérification de la perfusion de la tête du nerf optique immédiatement après l'injection et une tonométrie dans les 30 minutes suivant l'injection).
Examen ophtalmologique : l'administration de ce médicament peut entrainer une anomalie des examens ophtalmologiques réalisés. Ceux-ci comprennent la tomographie par cohérence optique (OCT), l'ophtalmoscopie (reflet fovéal), le test de vision des couleurs (test 28 Hue de Roth) et l'ERG plein champ.
CONTACTER LE MEDECIN/OPHTALMOLOGISTE IMMEDIATEMENT en cas de :
- Diminution sévère de la vision.
- Douleur oculaire, l'aggravation de la rougeur des yeux, une vision sévèrement trouble ou diminuée, une sensibilité accrue à la lumière ou une augmentation du nombre de points sombres flottants dans le champ de vision (corps flottants). Ils peuvent être les signes d'une infection, de saignement, de décollement ou de déchirure de la rétine ou d'une augmentation de la pression à l'intérieur de l'oeil traité.
- Une fluctuation de la vision, une vision double, des maux de tête, des halos lumineux, des nausées et vomissements.
Prudence en cas de conduite de véhicule ou machine : ce traitement peut entrainer des troubles visuels temporaires. Dans ces cas, les patients ne doivent ni conduire, ni utiliser des machines tant qu'ils n'ont pas récupéré une fonction visuelle suffisante).
Grossesse
Il n'existe pas de données sur l'utilisation de JETREA chez la femme enceinte. Aucune étude toxicologique sur la reproduction n'a été réalisée. Il est attendu que l'exposition systémique à JETREA soit très faible après une injection intravitréenne. JETREA ne doit être utilisé pendant la grossesse que si le bénéfice clinique l'emporte sur les risques potentiels.
Allaitement
On ne sait pas si JETREA est excrété dans le lait maternel. JETREA ne doit être utilisé au cours de l'allaitement que si le bénéfice clinique l'emporte sur les risques potentiels.
Fécondité
Il n'existe pas de données sur l'effet de JETREA sur la fécondité.
Aucune étude formelle d'interaction n'a été réalisée.
L'ocriplasmine est une enzyme protéolytique avec une activité de sérine protéase qui peut persister dans l'œil pendant plusieurs jours après l'injection intravitréenne (voir rubrique Propriétés pharmacocinétiques). L'administration à court intervalle en association avec d'autres médicaments dans le même œil peut affecter l'activité des deux médicaments et n'est donc pas recommandée.
Il n'existe pas de données cliniques sur l'usage concomitant de l'ocriplasmine avec les anti-VEGF (pour Vascular Endothelium Growth Factor) et il n'est donc pas recommandé.
Aucune interaction systémique n'est attendue.
JETREA doit être préparé et administré par un ophtalmologiste qualifié, expérimenté dans les injections intra-vitréennes. Le diagnostic de traction vitréo-maculaire (TVM) doit comprendre un tableau clinique complet dont les antécédents du patient, un examen clinique et des explorations utilisant des outils de diagnostic actuels, tels que la tomographie par cohérence optique (OCT).
Posologie
La dose recommandée est de 0,125 mg (0,1 mL de solution diluée), administrée par injection intra- vitréenne dans l'œil atteint, une seule fois, en dose unique. Chaque flacon ne doit être utilisé qu'une seule fois et pour le traitement d'un seul œil. Le traitement avec JETREA dans l'autre œil n'est pas recommandé immédiatement ou dans les 7 jours suivant l'injection initiale afin de contrôler le traitement après l'injection y compris le potentiel de baisse de vision de l'œil injecté. L'administration répétée dans le même œil n'est pas recommandée (voir rubrique Mises en garde et précautions d'emploi).
Voir la rubrique Mises en garde et précautions d'emploi pour les instructions sur le suivi après injection. Populations particulières
Insuffisance rénale
Aucune étude formelle n'a été menée avec JETREA chez des patients insuffisants rénaux. Aucun ajustement de la posologie, ni aucune restriction particulière ne sont prévus pour les patients insuffisants rénaux (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
Aucune étude formelle n'a été menée avec JETREA chez des patients insuffisants hépatiques. Aucun ajustement de la posologie, ni aucune restriction particulière ne sont prévus pour les patients insuffisants hépatiques (voir rubrique Propriétés pharmacocinétiques).
Sujets âgés
La population des personnes âgées a été étudiée dans des études cliniques. Aucun ajustement de la posologie n'est nécessaire.
Population pédiatrique
L'utilisation de JETREA dans l'indication de la traction vitréo-maculaire (TVM) n'est pas justifiée chez les enfants âgés de moins de 18 ans, y compris lorsqu'elle est associée à un trou maculaire d'un diamètre inférieur ou égal à 400 microns. Les données actuellement disponibles relatives à un usage pédiatrique sont décrites rubrique Propriétés pharmacodynamiques.
Mode d'administration
Flacon à usage unique réservé à la voie intravitréenne.
Un traitement préopératoire par collyre antibiotique pourra être administré à la discrétion de l'ophtalmologiste.
Précautions à prendre avant la manipulation ou l'administration du médicament
La procédure d'injection intravitréenne doit être menée dans des conditions aseptiques contrôlées, qui comprennent l'usage d'une désinfection chirurgicale des mains, de gants stériles, d'un drap stérile, d'un spéculum oculaire stérile (ou équivalent) et de la mise à disposition de paracentèse stérile (si nécessaire). La peau péri-oculaire, la paupière et la surface oculaire doivent être désinfectées, une anesthésie adéquate doit être pratiquée et un antiseptique topique à large spectre administré juste avant l'injection conformément aux pratiques médicales standard en vigueur.
Pour les instructions concernant la dilution du médicament avant administration, voir la rubrique Instructions pour l'utilisation, la manipulation et l'élimination.
L'aiguille d'injection doit être insérée 3,5-4,0 mm en arrière du limbe en se dirigeant vers le centre de la cavité vitréenne et en évitant le méridien horizontal. Le volume d'injection de 0,1 mL est alors administré au sein du corps vitré.
Durée de conservation :
3 ans lorsqu'il est conservé au congélateur (-20°C±5°C).
Après décongélation
Le médicament doit être dilué et utilisé immédiatement. Toutefois, la stabilité chimique et physique du produit non ouvert conservé dans son emballage d'origine à l'abri de la lumière a été démontrée pendant 8 heures à une température ne dépassant pas 25°C. Ne pas recongeler le flacon une fois qu'il a été décongelé.
Après ouverture/dilution
Du point de vue microbiologique, le médicament doit être utilisé immédiatement après ouverture/dilution. Le flacon et tout résidu inutilisé de la solution diluée doivent être jetés après une utilisation unique.
Précautions particulières de conservation :
A conserver au congélateur (-20°C±5°C).
Pour les conditions de conservation du médicament après décongélation et ouverture/dilution, voir la rubrique Durée de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments à l'exception d'une solution injectable stérile de chlorure de sodium 9 mg/mL (0,9%), sans conservateur, non tamponnée.
:
Inspecter visuellement le flacon pour déceler toute particule de matière. Seule une solution claire, incolore, et sans particule visible doit être utilisée.
Les données cliniques sur les effets du surdosage de JETREA sont limitées. Un cas de surdosage accidentel de 0,250 mg d'ocriplasmine (deux fois la dose recommandée) a été signalé. Le patient a présenté une baisse de la MAVC de 21 lettres ETDRS par rapport à la référence, qui est revenue à 9 lettres de la référence à la fin de l'étude. Le patient a également développé une légère hyperhémie
conjonctivale, une inflammation oculaire et un myosis, qui se sont résolus après un traitement à base de corticostéroïdes en collyre.
En cas de surdosage, il est recommandé de mettre en place une surveillance étroite. Si un effet indésirable se produit, il doit être traité conformément aux pratiques médicales standard.
Classe pharmacothérapeutique : Médicaments ophtalmologiques, autres médicaments ophtalmologiques, code ATC : S01XA22
Mécanisme d'action
L'ocriplasmine a une activité protéolytique contre les composants protéiques du corps vitré et de l'interface vitréo-rétinienne (IVR) (par ex., laminine, fibronectine et collagène), et vise à dissoudre la matrice protéique responsable de l'adhérence vitréo-maculaire anormale (AVM). L'étroite liaison des composants protéiques au sein de la zone maculaire de l'IVR contribue à la traction vitréo-maculaire (TVM), entraînant l'altération visuelle ou les trous maculaires.
Efficacité et sécurité clinique
L'efficacité et la sécurité clinique de JETREA pour le traitement de la traction vitréo-maculaire (TVM) ont été évaluées dans le cadre de 3 études menées en double aveugle.
Etudes TG-MV-006 et TG-MV-007
L'efficacité de JETREA a été démontrée dans deux études pivot multicentriques, randomisées, en double aveugle, contrôlées contre placebo, d'une durée de six mois chez des patients présentant une TVM. Au total, 652 patients (JETREA 464, placebo 188) ont été randomisés dans ces deux études.
Dans les deux études pivot, la proportion de patients ayant obtenu une résolution de l'AVM au jour 28 (le critère principal) a été significativement (p ≤ 0,003) plus élevée dans le groupe de JETREA comparativement au groupe du placebo. La différence a continué d'être statistiquement significative jusqu'au mois 6 dans chaque étude (p ≤ 0,024). D'après les données intégrées, 26,5% des patients dans le groupe de JETREA comparativement à 10,1% des patients dans le groupe du placebo, ont obtenu une résolution de l'AVM au jour 28 (p < 0,001). La différence statistique s'est maintenue du jour 7 au mois 6 (Figure 1).
Figure 1: Proportion des patients obtenant une résolution de l'AVM jusqu'au jour 180 (mois 6) (TG-MV-006, TG-MV-007 et données intégrées)
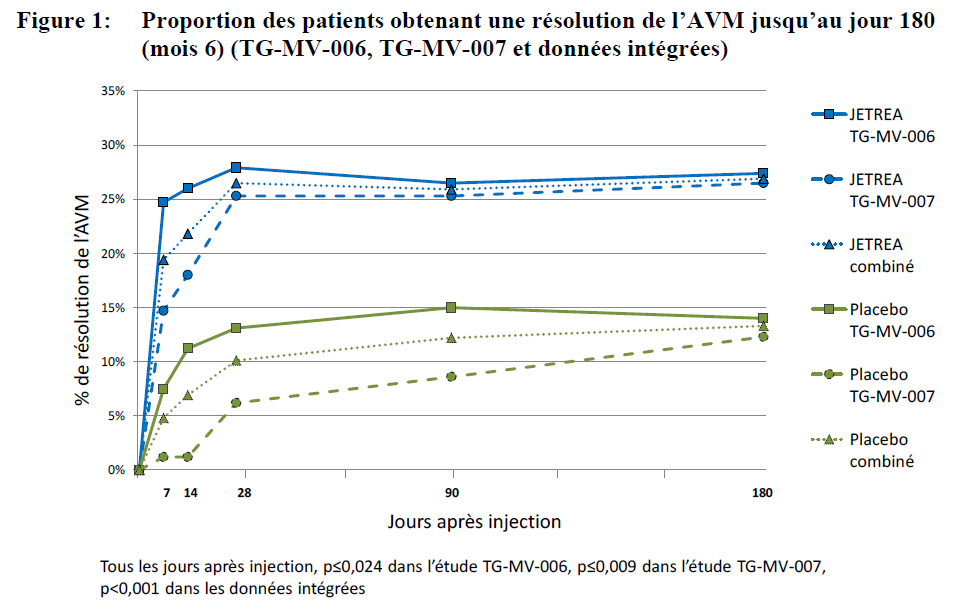
Les patients ne présentant pas de MER au départ étaient plus susceptibles d'obtenir une résolution de l'AVM au jour 28 par rapport à ceux présentant une MER au départ. D'après les données intégrées, le niveau de résolution de l'AVM au jour 28 était plus élevé chez les patients traités avec JETREA comparé au placebo dans les deux sous-groupes sans MER (37,4% contre 14,3%, p>0,001) et avec MER (8,7% contre 1,5%, p=0,046).
Les patients présentant un diamètre plus petit d'AVM au départ (≤ 1500 microns) étaient plus susceptibles d'obtenir une résolution de l'AVM au jour 28 par rapport à ceux présentant un diamètre > 1500 microns. D'après les données intégrées, le niveau de résolution de l'AVM au jour 28 était plus élevé chez les patients traités avec JETREA comparé au placebo dans les deux sous-groupes avec une AVM ≤ 1500 microns au départ (34,7% contre 14,6%, p<0,001) et avec une AVM > 1500 microns au départ (5,9% contre 0%, p=0,113).
D'après les données intégrées, un trou maculaire de pleine épaisseur (TMPE) était présent au début de l'étude chez 106/464 (22,8 %) patients et 47/188 (25 %) patients dans les groupes de JETREA et du placebo, respectivement. Parmi eux, la proportion de patients ayant obtenu la fermeture du trou maculaire de pleine épaisseur sans vitrectomie au jour 28 était plus élevée dans le groupe de JETREA que dans le groupe du placebo (40,6% contre 10,6%, respectivement ; p < 0,001). Une différence a été maintenue jusqu'à la fin des études (mois 6).
Un pourcentage significativement plus élevé de patients traités avec JETREA a présenté un décollement postérieur du vitré total (DPV) au jour 28 comparativement aux patients traités par placebo (données intégrées : 13,4% contre 3,7%, respectivement ; p < 0,001).
Au cours des études, la vitrectomie pouvait être effectuée à la discrétion de l'Investigateur. Les patients traités par JETREA étaient moins susceptibles d'avoir eu une vitrectomie à la fin de l'étude (mois 6) comparativement aux patients traités par placebo (données intégrées : 17,7% contre 26,6%, respectivement ; p = 0,016).
Une proportion plus élevée de patients traités par JETREA a gagné ≥ 2 ou ≥ 3 lignes lors de l'évaluation de la MAVC (indépendamment de la vitrectomie) au mois 6 (28,0% et 12,3%, respectivement) comparativement aux patients traités par placebo (17,1% et 6,4%) (p = 0,003 et p = 0,024, respectivement). Aussi la proportion de patients gagnant ≥ 2 ou ≥ 3 lignes lors de l'évaluation de la MAVC sans vitrectomie favorisait JETREA au mois 6 (23,7% contre 11,2%, p<0,001 pour un gain ≥ 2 lignes et 9,7% contre 3,7%, p=0,008 pour un gain ≥ 3 lignes).
Dans l'analyse intégrée du questionnaire VFQ-25 (National Eye Institute Visual Function Questionnaire-25), une différence numérique en faveur de JETREA par rapport au placebo a été démontrée dans chaque score de la sous-échelle, ainsi que dans le score composé. La différence d'amélioration dans le score de la sous-échelle relative à la vision globale était statistiquement significative (6,1 JETREA contre 2,1 placebo, p = 0,024).
Etude TG-MV-014
L'efficacité de JETREA a été confirmée dans une étude randomisée, en double aveugle, contrôlée contre injection simulée, d'une durée de 24 mois chez des patients présentant une TVM, finalisée après l'approbation initiale de l'autorisation de mise sur le marché. Au total, 220 patients (146 avec JETREA, 74 avec injection simulée) ont été randomisés dans cette étude.
La proportion de patients ayant obtenu une résolution de l'AVM au jour 28 (critère d'évaluation principal) était de 41,7% dans le groupe JETREA contre 6,2% dans le groupe injection simulée (p<0,001). Cet effet s'est maintenu dans le temps et le taux de résolution de l'AVM était systématiquement plus élevé dans le groupe JETREA à chaque visite de l'étude suivant l'injection par rapport au groupe injection simulée.
Dans cette étude, un TMPE était présent au début de l'étude chez 50/145 (34,5%) patients et 26/73 (35,6%) patients dans les groupes JETREA et injection simulée, respectivement. Parmi eux, 30% des patients traités par JETREA et 15,4% des patients du groupe injection simulée ont présenté une fermeture non chirurgicale du TMPE au mois 24. Il en était de même pour tous au mois 3.
La proportion de patients ayant eu recours à une vitrectomie était plus faible dans le groupe JETREA que dans le groupe injection simulée à toutes les visites. Au mois 24, les proportions étaient de 48/145 (33,3%) et 32/73 (43%), respectivement. La cause la plus fréquente de recours à une vitrectomie était le TMPE (chez 24,8% des patients traités par JETREA et 23,3% des patients ayant reçu une injection simulée). La proportion de patients ayant eu recours à une vitrectomie pour un événement d'AVM/de TVM était de 8,3% dans le groupe JETREA contre 19,2% dans le groupe injection simulée.
La proportion de patients ayant gagné ≥ 2 ou ≥ 3 lignes lors de l'évaluation de la MAVC au mois 6, indépendamment de la vitrectomie, était légèrement plus élevée dans le groupe JETREA (36,2%, 18,6%) que dans le groupe injection simulée (28,6%, 13,1%). Au mois 24, la proportion de patients ayant gagné ≥ 2 lignes par rapport au début de l'étude lors de l'évaluation de la MAVC était plus importante dans le groupe JETREA que dans le groupe injection simulée (50,5% contre 39,1%). La proportion de patients ayant gagné ≥ 3 lignes par rapport au début de l'étude était uniquement supérieure dans le groupe JETREA (23,4% contre 12,8%, respectivement), dans le sous-groupe qui ne présentait pas de TMPE au début de l'étude. Le gain de ≥ 2 ou ≥ 3 lignes lors de l'évaluation de la MAVC sans vitrectomie favorisait JETREA par rapport à l'injection simulée au mois 6 (26,8%, 14,0% contre 15,62%, 6,2%, respectivement) et au mois 24 (31,9%, 16,8% contre 11,7%, 4,1%, respectivement).
Une proportion plus importante de patients du groupe JETREA présentait une amélioration ≥ 5 points du score global et de chaque sous-échelle du questionnaire VFQ-25, indépendamment de la vitrectomie, à toutes les visites. Au mois 24, 51,4% des patients traités par JETREA présentait une amélioration ≥ 5 points du score global du questionnaire VFQ-25, contre 30,1% dans le groupe injection simulée.
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec JETREA dans tous les sous-groupes de la population pédiatrique dans le traitement de la traction vitréo-maculaire (TVM), notamment lorsqu'elle est associée à un troumaculaire d'un diamètre inférieur ou égal à 400 microns (voir rubrique Posologie et mode d'administration pour des informations concernant l'utilisation pédiatrique).
La tolérance et l'efficacité de l'ocriplasmine chez les sujets pédiatriques programmés pour une vitrectomie ont été étudiées dans l'étude TG-MV-009. Une dose de 0,175 mg d'ocriplasmine (dose supérieure à la dose recommandée) ou de placebo, a été administrée par injection intra-vitréenne unique dans 24 yeux d'enfants âgés de 0 à 16 ans, 30 à 60 minutes avant l'horaire programmé de la vitrectomie. Les principales causes justifiant la vitrectomie étaient un décollement de la rétine et une rétinopathie du prématuré. Le traitement avec l'ocriplasmine n'a pas démontré d'effet sur le taux de décollement postérieur du vitré, le stade de liquéfaction du vitré, le taux de ré-application immédiate post-opératoire de la rétine, le développement d'une vitréorétinopathie proliférante, ou le stade de la rétinopathie du prématuré. Les données de sécurité issues de l'étude TG-MV-009 sont cohérentes avec le profil de sécurité connu pour JETREA. Sur la base des résultats de cette étude, l'utilisation de JETREA comme complément de la vitrectomie chez les enfants, afin de faciliter la séparation et le retrait du vitré, n'est pas recommandée.
Origine ethnique
L'expérience est limitée dans les groupes autres que celui des caucasiens.
Les taux d'ocriplasmine dans le vitré diminuent rapidement après l'administration intravitréenne. Dans une étude clinique chez des patients devant subir une vitrectomie et recevant 0,125 mg de JETREA (correspondant à une concentration initiale théorique de 29 µg/mL dans le vitré), l'activité moyenne de l'ocriplasmine était de 9% de la concentration initiale théorique 2 à 4 heures après l'injection et en dessous du taux le plus faible de quantification au bout de 7 jours.
En raison de la petite dose administrée (0,125 mg), il n'est pas attendu que les taux d' ocriplasmine soient détectables dans la circulation systémique après une injection intravitréenne.
Lorsqu'elle est administrée par voie intraveineuse, l'ocriplasmine entre dans la voie du catabolisme des protéines endogènes dans laquelle elle est rapidement inactivée par ses interactions avec des inhibiteurs de la protéase α2-antiplasmine ou α2macroglobuline. Le complexe inactif d'ocriplasmine/ α2-antiplasmine est éliminé de la circulation avec une demi-vie (t1/2) de plusieurs heures.
Insuffisance rénale
Aucune étude n'a été menée pour examiner la pharmacocinétique de l'ocriplasmine chez des patients insuffisants rénaux étant donné qu'il est attendu que l'exposition systémique soit très faible après une administration intravitréenne.
Insuffisance hépatique
Aucune étude n'a été menée pour examiner la pharmacocinétique de l'ocriplasmine chez des patients insuffisants hépatiques étant donné qu'il est attendu que l'exposition systémique soit très faible après une administration intravitréenne.
L'injection intravitréenne de JETREA peut avoir une influence modérée sur l'aptitude à conduire des véhicules et à utiliser des machines en raison d'éventuels troubles visuels temporaires (voir
rubrique Effets indésirables). Dans ces cas, les patients ne doivent ni conduire, ni utiliser des machines tant qu'ils n'ont pas récupéré une fonction visuelle suffisante.
La toxicité intravitréenne de l'ocriplasmine a été évaluée chez le lapin, le singe et le mini porc. L'ocriplasmine a induit une réponse inflammatoire et des changements transitoires de l'ERG chez le lapin et le singe, alors qu'aucune inflammation ou aucun changement de l'ERG n'ont été observés chez le mini porc. Chez le lapin et le singe, l'incidence d'infiltrations de cellules vitréennes a eu tendance à se résorber dans le temps. Chez le singe, après l'administration de 125 µg/oeil (68 µg/mL dans le vitré), l'enregistrement de l'ERG est redevenu normal au jour 55. Une subluxation du cristallin a été observée chez les trois espèces à des concentrations d'ocriplasmine supérieures ou égales à 41 µg/mL dans le vitré, soit à une concentration supérieure à celle attendue chez l'homme, de 29 µg/mL. Cet effet semble être lié à la dose et a été observé chez tous les animaux ayant reçu plus d'une fois de l'ocriplasmine intravitréenne. Des changements pathologiques liés à l'hémorragie intraoculaire ont été observés chez le lapin et le singe. L'origine de cette hémorragie demeure inconnue et pourrait être liée à la procédure d'injection elle-même ou à l'administration d'ocriplasmine. Aucune toxicité systémique n'a été observée après l'administration intravitréenne d'ocriplasmine.
La toxicité systémique d'ocriplasmine a été évaluée chez le rat et le chien. L'administration intraveineuse de 10 mg/kg a été généralement bien tolérée chez le rat et chez le chien après administration d'une dose unique ou d'une dose répétée.
Aucune donnée de cancérogenèse, de mutagenèse ou de toxicité sur la reproduction ou le développement, n'est disponible.
Les flacons sont réservés à un usage unique.
Pour la préparation de JETREA pour administration intravitréenne, respectez les instructions suivantes :
Retirer le flacon du congélateur et laisser décongeler à température ambiante (cela prend environ 2 minutes).
Une fois entièrement décongelé, retirer le capuchon de protection orange amovible en polypropylène du flacon.

Désinfecter le haut du flacon à l'aide d'une lingette imbibée d'alcool.

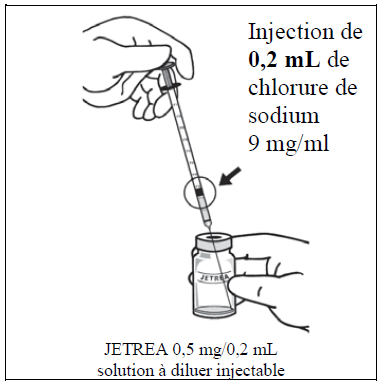
À l'aide d'une technique aseptique, diluer en ajoutant 0,2 mL d'une solution injectable de chlorure de sodium 9 mg/mL (0,9%) (stérile, sans conservateur, non tamponnée) dans le flacon de JETREA et remuer délicatement le flacon jusqu'à ce que les solutions soient mélangées. Le diluant doit être retiré d'un récipient non ouvert qui ne doit être utilisé qu'une fois. Le reste de la solution injectable de chlorure de sodium 9 mg/mL (0,9%) doit être jeté. La solution diluée doit être utilisée immédiatement.

Inspecter visuellement le flacon pour déceler toute particule de matière. Seule une solution claire, incolore, et sans particule visible doit être utilisée.
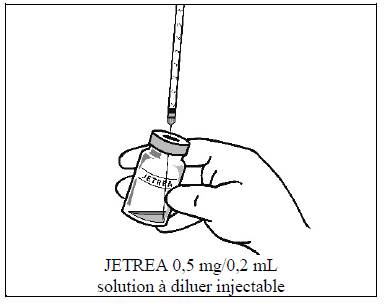
À l'aide d'une technique aseptique, prélever toute la solution diluée en utilisant une aiguille stérile appropriée (incliner légèrement le flacon pour faciliter le retrait) et jeter l'aiguille après avoir prélevé le contenu du flacon. Ne pas utiliser cette aiguille pour l'injection intravitréenne.
-
Remplacer l'aiguille par une aiguille stérile appropriée, expulser soigneusement de la seringue l'excès de volume en appuyant lentement sur le piston de sorte que la pointe du piston soit alignée avec le trait de la seringue marquant 0,1 mL (correspondant à 0,125 mg d'ocriplasmine).

Injecter 0,1 mL de solution immédiatement dans le milieu du corps vitré.
Jeter le flacon et toute portion inutilisée de la solution après une utilisation unique.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Médicament réservé à l’usage hospitalier.
Prescription réservée aux spécialistes en ophtalmologie.
Solution à diluer injectable (solution à diluer stérile). Solution claire et incolore.
Solution de 0,2 mL dans un flacon (verre de type I) fermé par un bouchon en caoutchouc chlorobutyle et un capuchon amovible orange en polypropylène. Emballage contenant 1 flacon.