
BYDUREON 2 mg, poudre et solvant pour suspension injectable à libération prolongée en stylo prérempli, boîte de 4 stylos préremplis de 2 mg
Retiré du marché le : 05/10/2022
Dernière révision : 11/01/2021
Taux de TVA : 10%
Laboratoire exploitant : ASTRAZENECA
Source :

Bydureon est indiqué chez les adultes âgés de 18 ans ou plus atteints de diabète de type 2 pour améliorer le contrôle de la glycémie en association avec d'autres médicaments destinés au traitement du diabète incluant l'insuline basale, lorsque le traitement en cours, en complément d'un régime alimentaire et d'une activité physique, ne permet pas d'assurer un contrôle adéquat de la glycémie.
Pour les résultats des études concernant les associations, les effets sur le contrôle glycémique et les événements cardiovasculaires, et les populations étudiées, voir les rubriques Mises en garde spéciales et précautions d'emploi, Interactions avec d'autres médicaments et autres formes d'interactions et Propriétés pharmacodynamiques.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
L'exénatide à libération prolongée ne doit pas être utilisé
chez les patients ayant un diabète de type 1 ou une acidocétose diabétique.
L'exénatide à libération prolongée ne peut pas se
substituer à l'insuline. Des cas d'acidocétose diabétique ont été rapportés
chez des patients insulino-dépendants après une interruption rapide de la prise
d'insuline ou une réduction rapide de la dose d'insuline (voir rubrique Posologie
et mode d'administration).
L'exénatide à libération prolongée ne doit pas être administré par voie intraveineuse ou intramusculaire.
Insuffisance
rénale
Chez les
patients ayant une insuffisance rénale terminale dialysés, la fréquence et la
sévérité des effets indésirables gastro-intestinaux sont augmentées par des
doses uniques d'exénatide à libération immédiate, par
conséquent l'exénatide à libération prolongée n'est
pas recommandé chez les patients ayant une insuffisance rénale terminale ou une
atteinte sévère de la fonction rénale (DFG < 30 mL/min).
Il y a eu des notifications peu fréquentes d'altération de la fonction rénale avec l'exénatide, incluant des cas d'augmentation de la créatinine sérique, d'atteinte rénale, d'aggravation d'une insuffisance rénale chronique et d'insuffisance rénale aiguë, nécessitant parfois une hémodialyse. Certains de ces évènements sont survenus chez des patients qui présentaient par ailleurs d'autres conditions pouvant entraîner une déshydratation parmi lesquelles des nausées, des vomissements et/ou des diarrhées et/ou recevant des médicaments connus pour affecter la fonction rénale et l'état d'hydratation. Ces médicaments peuvent être : les inhibiteurs de l'enzyme de conversion, les antagonistes de l'angiotensine II, les médicaments anti-inflammatoires non-stéroïdiens et les diurétiques. L'altération de la fonction rénale a été réversible sous traitement symptomatique et après l'arrêt des médicaments potentiellement en cause, dont l'exénatide.
Maladie
gastro-intestinale sévère
L'exénatide à libération prolongée n'a pas été étudié chez
les patients ayant une pathologie gastro- intestinale sévère, dont la gastroparésie. Son utilisation est souvent associée à des
effets indésirables gastro-intestinaux incluant des nausées, des vomissements
et des diarrhées. L'utilisation de l'exénatide à
libération prolongée n'est donc pas recommandée chez les patients atteints
d'une maladie gastro- intestinale sévère.
Pancréatite
aiguë
L'utilisation
des agonistes du récepteur GLP-1 a été associée à un risque de développement de
pancréatites aiguës. Dans les études cliniques portant sur l'exénatide à libération prolongée, une pancréatite aiguë est
survenue chez 0,3 % des patients. Il y a eu des notifications spontanées de
pancréatites aiguës avec l'exénatide à libération
prolongée. L'évolution des pancréatites a été favorable sous traitement
symptomatique, à l'exception de très rares cas de pancréatite nécrosante ou
hémorragique et/ou de décès rapportés. Les patients doivent être informés des
symptômes caractéristiques des pancréatites aiguës : une douleur abdominale
sévère et persistante. Si une pancréatite est suspectée, l'exénatide
à libération prolongée doit être arrêté ; si la pancréatite aiguë est
confirmée, l'exénatide à libération prolongée ne doit
pas être réadministré. La prudence s'impose chez les
patients avec des antécédents de pancréatite.
Association
de médicaments
L'utilisation
de l'exénatide à libération prolongée en association
avec les dérivés de la D- phénylalanine (les méglitinides),
les inhibiteurs de l'alpha-glucosidase, les
inhibiteurs de la dipeptidyl peptidase-4 ou les
agonistes des récepteurs au GLP-1, n'a pas été étudiée. L'utilisation de l'exénatide à libération prolongée en association à l'exénatide à libération immédiate n'a pas été étudiée et
n'est pas recommandée.
Interaction
avec la warfarine
Des cas
d'augmentation de l'INR (International Normalized
Ratio) ont été spontanément rapportés, parfois associées à des saignements,
lors de l'utilisation concomitante de la warfarine et
de l'exénatide (voir rubrique Interactions avec
d'autres médicaments et autres formes d'interactions).
Hypoglycémie
Le risque
d'hypoglycémie était augmenté lorsque l'exénatide à
libération prolongée était utilisé en association à un sulfamide hypoglycémiant
au cours des études cliniques. En outre, dans les études cliniques, l'incidence
des hypoglycémies était augmentée chez les patients ayant une insuffisance
rénale légère et traités par une association comportant un sulfamide
hypoglycémiant, par rapport aux patients ayant une fonction rénale normale.
Afin de diminuer le risque d'hypoglycémie associé à l'utilisation d'un
sulfamide hypoglycémiant, une diminution de la dose du sulfamide hypoglycémiant
doit être envisagée.
Perte de
poids rapide
Une perte de
poids rapide supérieure à 1,5 kg par semaine a été observée chez des patients
traités par l'exénatide. Une perte de poids de cette
importance pourrait avoir des conséquences délétères. Les patients ayant une
perte de poids rapide doivent être surveillés à la recherche de signes et de
symptômes de cholélithiase.
Arrêt de
traitement
Après l'arrêt
du traitement, l'effet de l'exénatide à libération
prolongée peut perdurer car les taux plasmatiques d'exénatide
diminuent pendant plus de 10 semaines. Par conséquent, le choix d'autres médicaments
et de leur dose doit être pris en compte, car des effets indésirables peuvent
continuer à se produire et l'efficacité peut, au moins en partie, persister
tant que les taux d'exénatide diminuent.
Excipients
Contenu en
sodium : ce médicament contient moins d'1 mmol de
sodium (23 mg) par dose, et peut donc être considéré comme pratiquement « sans
sodium ».
Résumé du
profil de sécurité d'emploi
Les effets
indésirables les plus fréquents étaient principalement gastro-intestinaux
(nausées qui étaient l'effet indésirable le plus fréquent et qui étaient
associées à l'initiation du traitement et qui diminuaient avec le temps, et des
diarrhées). Par ailleurs, des réactions au site d'injection (prurits, nodules,
érythèmes), une hypoglycémie (avec les sulfamides hypoglycémiants), et des
céphalées ont été observées. La plupart des effets indésirables associés à
l'utilisation de l'exénatide à libération prolongée
étaient d'intensité légère à modérée.
Tableau
récapitulatif des effets indésirables
Les fréquences
des effets indésirables de l'exénatide à libération
prolongée identifiés à partir des études cliniques et des notifications
spontanées (non observés dans les études cliniques, fréquence indéterminée)
sont résumées dans le Tableau 1 ci-dessous.
Dans les études cliniques de l'exénatide à libération prolongée, les traitements de base incluaient un régime alimentaire et une activité physique, la metformine, un sulfamide hypoglycémiant, une thiazolidinedione, une association de traitements hypogycémiants oraux ou une insuline basale.
Les effets indésirables sont listés ci-dessous selon la terminologie MedDRA par classe de système d'organe et par fréquence. Les fréquences sont définies de la manière suivante : très fréquent (≥ 1/10), fréquent (≥ 1/100, < 1/10), peu fréquent (≥ 1/1 000, < 1/100), rare (≥ 1/10 000, < 1/1 000), très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 1 : Effets indésirables de l'exénatide à libération prolongée identifiés dans les études cliniques et les notifications spontanées
|
Classe de système d'organes /Effets indésirables |
Fréquence de survenue |
|||||
|
Très fréquent |
Fréquent |
Peu fréquent |
Rare |
Très rare |
Fréquence indéterminée |
|
|
Affections hématologiques et du système lymphatique |
||||||
|
Thrombocytopénie d'origine médicamenteuse |
X4 |
|||||
|
Affections du système immunitaire |
||||||
|
Réaction anaphylactique |
X1 |
|||||
|
Troubles du métabolisme et de la nutrition |
||||||
|
Hypoglycémie (avec |
X1 |
|||||
|
un sulfamide hypoglycémiant) |
||||||
|
Hypoglycémie (avec insuline) |
X2, 3 |
|||||
|
Diminution de l'appétit |
X1 |
|||||
|
Déshydratation |
X1 |
|||||
|
Affections du système nerveux |
||||||
|
Céphalées |
X1 |
|||||
|
Sensation vertigineuse |
X1 |
|||||
|
Dysgueusie |
X1 |
|||||
|
Somnolence |
X1 |
|||||
|
Affections gastro- intestinales |
||||||
|
Obstruction intestinale |
X1 |
|||||
|
Pancréatite aiguë (voir rubrique Mises en garde spéciales et précautions d'emploi) |
X1 |
|||||
|
Nausées |
X1 |
|||||
|
Vomissements |
X1 |
|||||
|
Diarrhée |
X1 |
|||||
|
Dyspepsie |
X1 |
|||||
|
Douleur abdominale |
X1 |
|||||
|
Reflux gastro- oesophagien |
X1 |
|||||
|
Distension abdominale |
X1 |
|||||
|
Eructation |
X1 |
|||||
|
Constipation |
X1 |
|||||
|
Flatulence |
X1 |
|||||
|
Retard de la vidange gastrique |
X5 |
|||||
|
Affections de la peau et du tissu sous- cutané |
||||||
|
Eruption maculaire ou papuleuse |
X4 |
|||||
|
Prurit, et / ou urticaire |
X1 |
|||||
|
Oedème angioneurotique |
X4 |
|||||
|
Abcès et cellulite au site d'injection |
X4 |
|||||
|
Hyperhidrose |
X1 |
|||||
|
Alopécie |
X1 |
|||||
|
Affections du rein et des voies urinaires |
||||||
|
Altération de la fonction rénale incluant insuffisance rénale aiguë, aggravation d'une insuffisance rénale chronique, atteinte rénale, augmentation de la créatinine sérique |
X1 |
|||||
|
(voir rubrique Mises en garde spéciales et précautions d'emploi). |
||||||
|
Troubles généraux et anomalies au site d'administration |
||||||
|
Prurit au site d'injection |
X1 |
|||||
|
Fatigue |
X1 |
|||||
|
Erythème au site d'injection |
X1 |
|||||
|
Eruption au site d'injection |
X1 |
|||||
|
Asthénie |
X1 |
|||||
|
Sensation de nervosité |
X1 |
|||||
|
Investigations |
||||||
|
INR augmenté (voir rubrique Mises en garde spéciales et précautions d'emploi) |
X4 |
|||||
1 Fréquence établie à partir de la base de données des douze études à long terme terminées d'efficacité et de sécurité de l'exénatide à libération prolongée. N total = 2868, (patients sous sulfamide hypoglycémiant n = 1002)
2 Basée sur les événements d'hypoglycémie qui :
1. résultent d'une perte de conscience, de convulsions, ou d'un coma qui se sont résolus après administration de glucagon ou de glucose OU
2. demandent l'assistance d'une tierce personne pour la prise en charge du fait d'une altération de la conscience ou du comportement et présentant une glycémie < 54 mg/dL (3 mmol/L) OU
3. présentent des symptômes d'une hypoglycémie avec une glycémie concomitante < 54 mg/dL (3 mmol/L) avant traitement.
3 Fréquence rapportée, de la période de traitement de 28 semaines de l'étude contrôlée, de l'exénatide à libération prolongée en ajout de l'insuline glargine (N=231).
4 Fréquence établie à partir des données issues des notifications spontanées de l'exénatide à libération prolongée (dénominateur inconnu).
5 Fréquence établie à partir de 16 études d'efficacité et de sécurité à long terme réalisées avec l'exénatide à libération prolongée N = 4086 au total.
Description
des effets indésirables sélectionnés
Thrombocytopénie d'origine
médicamenteuse
Une thrombocytopénie d'origine médicamenteuse, avec des
anticorps anti-plaquettaires induits par l'exénatide, a été signalé dans le cadre du suivi
post-commercialisation. La thrombocytopénie d'origine
médicamenteuse est une réaction à médiation immunitaire qui est causée par des
anticorps, médicament dépendant, ciblant les plaquettes. Ces anticorps
entrainent une destruction des plaquettes en présence du médicament
sensibilisant.
Hypoglycémie
L'incidence
des hypoglycémies était augmentée quand l'exénatide à
libération prolongée était associé à un sulfamide hypoglycémiant (24,0 % versus
5,4 %) (voir rubrique Mises en garde spéciales et précautions d'emploi).
Afin de réduire le risque d'hypoglycémie associé à l'utilisation d'un sulfamide
hypoglycémiant, une réduction de la dose de sulfamide hypoglycémiant peut être
envisagée (voir rubriques Posologie et mode d'administration et Mises
en garde spéciales et précautions d'emploi).
L'exénatide à libération prolongée était associé à une incidence des épisodes d'hypoglycémie significativement plus faible que l'insuline basale chez les patients recevant également un traitement par metformine (3 % versus 19 %) et chez les patients recevant également un traitement par metformine plus sulfamide hypoglycémiant (20 % versus 42 %).
A travers 12 études, la plupart des épisodes (99,9 % n=649) d'hypoglycémie étaient mineurs, et résolus avec une administration orale d'hydrate de carbone. Une hypoglycémie majeure a été rapportée chez un patient qui a eu une glycémie faible (2,2 mmol/L) et a nécessité une assistance avec un traitement oral par hydrate de carbone qui a résolu l'effet indésirable.
Quand l'exénatide à libération prolongée était ajouté à l'insuline basale, aucun ajustement de la dose initiale d'insuline ne s'est avéré nécessaire. L'exénatide à libération prolongée en association avec l'insuline basale n'a montré aucune différence cliniquement significative de l'incidence des épisodes d'hypoglycémie comparée à l'insuline. Aucun épisode d'hypoglycémie majeure n'a été rapporté dans le groupe de l'exénatide à libération prolongé associé à l'insuline.
Nausées
L'effet
indésirable rapporté le plus fréquemment était des nausées. D'une façon
générale, 20 % des patients traités avec l'exénatide
à libération prolongée ont présenté au moins un épisode de nausées comparé à 34
% des patients traités avec l'exénatide à libération
immédiate. La plupart des épisodes de nausées étaient d'intensité légère à
modérée. Chez la plupart des patients ayant présenté des nausées lors de
l'initiation du traitement, la fréquence des nausées a diminué avec la
poursuite du traitement.
Dans l'étude contrôlée sur 30 semaines, l'incidence des sorties d'études pour effets indésirables était de 6 % chez les patients traités par l'exénatide à libération prolongée, de 5 % chez les patients traités par l'exénatide à libération immédiate. Dans les différents groupes de traitement, les effets indésirables ayant le plus fréquemment conduit à une sortie d'étude étaient des nausées et des vomissements. Les sorties d'étude liées aux nausées ou vomissements concernaient respectivement < 1 % des patients traités par l'exénatide à libération prolongée et 1 % des patients traités par l'exénatide à libération immédiate.
Réactions
au site d'injection
Des réactions
au site d'injection ont été observées plus fréquemment chez les patients
traités par l'exénatide à libération prolongée
comparé aux patients traités par un comparateur (16 % versus 2 à 7 %)
durant la phase contrôlée de 6 mois des études. Ces réactions au site
d'injection ont généralement été d'intensité légère et n'ont d'ordinaire pas
conduit à une sortie d'étude. Les patients peuvent recevoir un traitement
symptomatique pour les soulager, tout en continuant le traitement. Un autre
site d'injection doit être utilisé chaque semaine pour les injections
ultérieures. En post-commercialisation, des cas d'abcès et de cellulite au site
d'injection ont été rapportés.
Des petits nodules sous-cutanés au site d'injection ont été observés très fréquemment au cours des études cliniques, ce qui est cohérent avec les propriétés connues des formulations poly (D,L-lactide- co-glycolide) des microsphères de polymère. La plupart de ces nodules étaient asymptomatiques, n'influaient pas sur la participation à l'étude et disparaissaient au bout de 4 à 8 semaines.
Immunogénicité
Compte tenu
des propriétés potentiellement immunogènes des protéines et des peptides, les
patients traités par l'exénatide à libération prolongée
peuvent développer des anticorps anti-exénatide. Chez
la plupart des patients développant des anticorps, le taux d'anticorps a
diminué au cours du temps.
La présence d'anticorps (taux élevés ou faibles) ne prédit en rien le contrôle glycémique pour un patient donné.
Dans les études cliniques avec l'exénatide à libération prolongée, approximativement 45 % des patients avaient un faible taux d'anticorps anti-exénatide à la fin de l'étude. Globalement le pourcentage de patients avec anticorps était homogène à travers les études cliniques. Globalement, le contrôle glycémique (HbA1c) était comparable à celui observé chez les patients sans anticorps. En moyenne dans les études de phase 3, 12 % des patients avaient un taux plus élevé d'anticorps. La réponse glycémique à l'exénatide à libération prolongée était absente à la fin de la période contrôlée des études pour une partie d'entre eux ; 2,6 % des patients avec un taux élevé d'anticorps n'ont pas eu d'amélioration de la glycémie alors que 1,6 % des patients sans anticorps n'ont pas non plus présenté d'amélioration de la glycémie.
Les patients avec anticorps anti-exénatide ont tendance à présenter plus de réactions au site d'injection (par exemple : rougeur de la peau et démangeaison), en revanche, les taux et les types d'effets indésirables étaient similaires à ceux observés chez les patients sans anticorps anti-exénatide.
Au cours de l'étude de 30 semaines et des deux études de 26 semaines, l'incidence des réactions au site d'injection potentiellement immunogènes (le plus souvent prurit avec ou sans érythème) était de 9 % chez les patients traités par l'exénatide à libération prolongée. Ces réactions étaient moins fréquemment observées chez les patients sans anticorps (4 %) comparé aux patients avec anticorps (13 %), avec une incidence plus grande chez ceux avec un taux d'anticorps plus élevé.
L'étude d'échantillons sanguins avec anticorps anti-exénatide n'a montré aucune réaction croisée significative avec des peptides endogènes similaires (glucagon ou GLP-1).
Perte de
poids rapide
Dans une étude
à 30 semaines, approximativement 3 % (n=4/148) des patients traités par l'exénatide à libération prolongée ont présenté au moins une
période de perte de poids rapide (perte de poids supérieure à 1,5 kg/semaine
enregistrée entre deux visites d'étude consécutives).
Augmentation
de la fréquence cardiaque
Une
augmentation moyenne de la fréquence cardiaque (FC) de 2,6 battements par
minute (bpm) par rapport à la valeur initiale (74 bpm) a été observée lors de l'analyse poolée
des études cliniques avec l'exénatide à libération
prolongée. Quinze pour cent des patients traités par l'exénatide
à libération prolongée ont présenté des augmentations moyennes de la FC ≥
10 bpm ; environ 5% à 10% des patients au sein des
autres groupes de traitement ont présenté des augmentations moyennes de la FC ≥
10 bpm.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après
autorisation du médicament est importante. Elle permet une surveillance
continue du rapport bénéfice/risque du médicament. Les professionnels de santé
déclarent tout effet indésirable suspecté via le système national de
déclaration - voir Annexe V.
AUTOSURVEILLANCE : Une autosurveillance glycémique est nécessaire afin d'ajuster la dose des sulfamides hypoglycémiants ou d'insuline, notamment lors de l'instauration du traitement par exénatide à libération prolongée et de la réduction de l'insuline. L'adoption d'une approche par étapes de la réduction de la dose d'insuline est recommandée.
NE
PAS UTILISER
l'exénatide à
libération prolongée chez
les patients ayant un diabète de type 1 ou une acidocétose diabétique.
L'exénatide à
libération prolongée ne peut pas se substituer à l'insuline. Des cas
d'acidocétose diabétique ont été rapportés chez des patients insulino-dépendants
après une interruption rapide de la prise d'insuline ou une réduction rapide de
la dose d'insuline.
ARRETER
le traitement et CONSULTER un médecin en cas de douleurs importantes et
persistantes au niveau de l'estomac, avec ou sans vomissements.
CONSULTER
immédiatement un MEDECIN en cas de gonflement du visage, de la langue
ou de la gorge, de difficultés à avaler, d'urticaire et de difficultés
à respirer.
INFORMER le médecin en cas de perte de poids rapide (plus de 1,5 kg/semaine).
FEMME désirant une grossesse : arrêter le traitement au moins 3 mois avant la grossesse planifiée.
PRUDENCE
en cas de conduite de véhicule ou d'utilisation de machine lors de
l'association avec un médicament sulfamide hypoglycémiant (risque
d'hypoglycémie).
OUBLI d'une dose :
- En cas d'oubli d'une dose, celle-ci doit être administrée dès que possible, à condition que la dose suivante soit prévue au moins 3 jours plus tard. Les patients peuvent ensuite reprendre l'administration hebdomadaire habituelle.
- En cas d'oubli d'une dose et la dose suivante étant prévue 1 ou 2 jours plus tard, le patient ne doit pas s'administrer la dose oubliée et doit reprendre l'administration de l'exénatide à libération prolongée le jour suivant prévu.
Femmes en âge d'avoir des enfants
En raison de la longue période d'élimination de l'exénatide à
libération prolongée, les femmes en âge de procréer doivent utiliser
une contraception pendant le traitement par l'exénatide à libération
prolongée. Ce médicament devra être interrompu au moins 3 mois avant
une grossesse planifiée.
Grossesse
Il n'existe pas de données suffisamment pertinentes concernant
l'utilisation de l'exénatide à libération prolongée chez la femme
enceinte. Des études effectuées chez l'animal ont mis en évidence une
toxicité sur la reproduction (voir rubrique Données de sécurité précliniques). Le risque potentiel chez l'homme n'est pas connu.
L'exénatide à libération prolongée ne doit pas être utilisé pendant la
grossesse et l'utilisation d'insuline est alors recommandée.
Allaitement
Aucune donnée n'existe sur l'excrétion de l'exénatide dans le lait
humain. L'exénatide à libération prolongée ne doit pas être utilisé
pendant l'allaitement.
Fécondité
Aucune étude de fécondité n'a été conduite chez l'homme.
Sulfamides hypoglycémiants
Un
ajustement de la dose du sulfamide hypoglycémiant peut être nécessaire du fait
de l'augmentation du risque d'hypoglycémie associée à un traitement par
sulfamide hypoglycémiant (voir rubriques Posologie et mode d'administration
et Mises en garde spéciales et précautions d'emploi).
Vidange
gastrique
Les résultats
d'une étude utilisant le paracétamol comme marqueur de la vidange gastrique
suggèrent que l'effet de l'exénatide à libération prolongée en tant que
ralentisseur de la vidange gastrique est mineur et qu'il n'est pas attendu que
cet effet entraîne des réductions cliniquement significatives du taux et de
l'étendue de l'absorption de traitements oraux administrés en association. Par
conséquent, aucun ajustement posologique n'est nécessaire pour les médicaments
sensibles à une vidange gastrique retardée.
Lorsque 1 000 mg de paracétamol en comprimés ont été administrés, avec ou sans repas, après 14 semaines de traitement par l'exénatide à libération prolongée, aucun changement significatif de l'ASC du paracétamol n'a été observé par rapport à la période contrôle. La Cmax du paracétamol diminuait de 16 % (à jeun) et de 5 % (post-prandial) et le tmax était augmenté approximativement d'1 heure à 1,4 heure (à jeun) et 1,3 heure (post-prandial) au cours de la période contrôle.
Les études d'interaction suivantes ont été conduites en utilisant 10 µg d'exénatide à libération immédiate mais pas avec exénatide à libération prolongée.
Warfarine
Un allongement
du tmax d'environ 2 h a été observé quand la warfarine a été administrée 35 min
après l'exénatide à libération immédiate. Aucun effet cliniquement significatif
n'a été observé sur la Cmax ou l'ASC. L'augmentation de l'INR a été
spontanément rapportée en cas d'association de la warfarine et de l'exénatide à
libération prolongée. Chez les patients traités par warfarine et/ou des dérivés
de la coumarine, l'INR doit être étroitement surveillé lors de l'initiation du
traitement par l'exénatide à libération prolongée (voir rubriques Mises en
garde spéciales et précautions d'emploi et Effets indésirables).
Inhibiteurs
de la hydroxy méthyl glutaryl-coenzyme A réductase
Quand
l'exénatide à libération immédiate était associé à une dose unique de
lovastatine (40 mg), l'ASC et la Cmax de la lovastatine étaient respectivement
diminuées d'environ 40 % et 28 %, le tmax étant allongé d'environ 4 heures par
rapport à la lovastatine administrée seule. Des études cliniques avec
l'exénatide à libération immédiate contrôlées versus placebo d'une durée
de 30 semaines ont montré que l'utilisation concomitante de l'exénatide et des
inhibiteurs de la HMG-CoA réductase n'était pas associée à des modifications
significatives des paramètres lipidiques (voir rubrique Propriétés pharmacodynamiques).
Aucune modification de posologie n'est nécessaire ; cependant les paramètres
lipidiques doivent être surveillés de façon appropriée.
Digoxine et
lisinopril
Il n'y a eu
aucun effet cliniquement significatif sur la Cmax ou l'ASC au cours des études
d'interaction de l'effet d'l'exénatide à libération immédiate sur la digoxine
ou le lisinopril, cependant un allongement du tmax d'environ 2 h a été observé.
Ethinyl
estradiol et lévonorgestrel
L'administration
d'une association de contraceptifs oraux (30 µg d'éthinyl estradiol et 150 µg
de lévonorgestrel) une heure avant l'exénatide à libération immédiate n'a pas
modifié l'ASC, la Cmax et la Cmin de l'éthinyl estradiol et du lévonorgestrel.
L'administration du contraceptif oral 35 min après l'exénatide n'a pas modifié
l'ASC mais a induit une diminution de la Cmax de l'ethinyl estradiol de 45 % et
de la Cmax du lévonorgestrel de 27-41 % ainsi qu'un retard de la tmax de 2-4 h
du fait du ralentissement de la vidange gastrique. La significativité clinique
de la diminution de la Cmax est limitée et aucun ajustement de dose des
contraceptifs oraux n'est nécessaire.
Population
pédiatrique
Les études
d'interaction avec l'exénatide n'ont été réalisées que chez l'adulte.
Posologie
La dose
recommandée est de 2 mg d'exénatide une fois par
semaine.
Chez les patients passant du traitement par l'exénatide à libération immédiate (Byetta) à l'exénatide à libération prolongée (Bydureon ou Bydureon BCise), il peut être observé des augmentations transitoires de la glycémie. La situation s'améliore généralement dans les deux premières semaines qui suivent l'initiation du traitement. Il est possible de passer d'un produit à base d'exénatide à libération prolongée à l'autre (Bydureon ou Bydureon BCise), sans qu'aucun effet significatif sur la glycémie soit attendu.
Quand l'exénatide à libération prolongée est associé à un traitement par metformine et/ou une thiazolidinedione, le traitement par metformine et/ou une thiazolidinedione peut être poursuivi à la même posologie. Quand il est associé à un traitement par un sulfamide hypoglycémiant, une diminution de la posologie du sulfamide hypoglycémiant doit être envisagée afin de diminuer le risque d'hypoglycémie (voir rubrique Mises en garde spéciales et précautions d'emploi).
L'exénatide à libération prolongée doit être administré une fois par semaine, le même jour chaque semaine. Le jour de l'administration hebdomadaire peut être modifié si nécessaire à condition que la dernière dose ait été administrée au moins trois jours avant. L'exénatide à libération prolongée peut être administré à n'importe quel moment de la journée, avec ou sans repas.
En cas d'oubli d'une dose, celle-ci doit être administrée dès que possible, à condition que la dose suivante soit prévue au moins 3 jours plus tard. Les patients peuvent ensuite reprendre l'administration hebdomadaire habituelle.
En cas d'oubli d'une dose et la dose suivante étant prévue 1 ou 2 jours plus tard, le patient ne doit pas s'administrer la dose oubliée et doit reprendre l'administration de l'exénatide à libération prolongée le jour suivant prévu.
L'utilisation de l'exénatide à libération prolongée ne nécessite pas d'autosurveillance supplémentaire. Une autosurveillance glycémique est nécessaire afin d'ajuster la dose des sulfamides hypoglycémiants ou d'insuline, notamment lors de l'instauration du traitement par exénatide à libération prolongée et de la réduction de l'insuline. L'adoption d'une approche par étapes de la réduction de la dose d'insuline est recommandée.
Si un traitement hypoglycémiant différent est initié après l'arrêt de l'exénatide à libération prolongée, la libération prolongée du produit doit être prise en compte (voir rubrique Propriétés pharmacocinétiques).
Populations particulières
Sujets âgés
Aucun
ajustement posologique n'est nécessaire en fonction de l'âge. Cependant, la
fonction rénale du patient doit être prise en compte car elle diminue
généralement avec l'âge (voir Atteinte de la fonction rénale) (voir
rubrique Propriétés pharmacocinétiques).
Atteinte de la fonction rénale
Aucun
ajustement posologique n'est nécessaire chez les patients avec une atteinte
légère ou modérée de la fonction rénale.
L'exénatide à libération prolongée n'est pas recommandé chez les patients avec une pathologie rénale terminale ou une atteinte sévère de la fonction rénale (débit de filtration glomérulaire [DFG] < 30 mL/min) (voir rubrique Mises en garde spéciales et précautions d'emploi).
Atteinte de la fonction hépatique
Aucun
ajustement posologique n'est nécessaire chez les patients avec une atteinte de
la fonction hépatique (voir rubrique Propriétés pharmacocinétiques).
Population
pédiatrique
La sécurité et
l'efficacité de l'exénatide à libération prolongée
chez les enfants et adolescents âgés de moins de 18 ans n'ont pas encore été
établies (voir rubrique Propriétés pharmacocinétiques). Les données
actuellement disponibles sont décrites dans la rubrique Propriétés
pharmacocinétiques mais aucune recommandation sur une posologie ne peut
être faite.
Mode
d'administration
Voie sous-cutanée
L'exénatide à libération prolongée est à administrer par le
patient lui-même. Chaque stylo peut uniquement être utilisé par une personne et
une seule fois.
Avant l'initiation d'exénatide à libération prolongée, il est fortement recommandé que les patients et les soignants soient forrmés par leur professionnel de santé. Le « Manuel d'utilisation », fourni à l'intérieur de la boîte, doit être suivi attentivement.
Chaque dose doit être administrée par injection sous-cutanée dans l'abdomen, la cuisse, ou l'arrière du bras immédiatement après la mise en suspension de la poudre dans le solvant.
Quand il est associé à de l'insuline, l'exénatide à libération prolongée et l'insuline doivent être administrés en deux injections séparées.
Pour les instructions concernant la mise en suspension du médicament avant administration, voir la rubrique Précautions particulières d'élimination et de manipulation et le « Manuel d'utilisation ».
Durée de conservation :
3 ans.
Après mise en suspension
La suspension doit être injectée immédiatement après le mélange de la poudre et du solvant.
Précautions particulières de conservation :
A conserver au réfrigérateur (entre 2 °C et 8 °C).
Ne pas congeler.
Les stylos peuvent être gardés jusqu'à 4 semaines à une température ne
dépassant pas 30 °C avant utilisation. A la fin de cette période, les
stylos doivent être utilisés ou jetés.
A conserver dans l'emballage extérieur d'origine afin de le protéger de la lumière.
Pour les conditions de conservation après reconstitution du médicament, voir la rubrique Durée de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Les effets de surdosages avec l'exénatide (fondés sur les études cliniques avec l'exénatide à libération immédiate) comportaient des nausées importantes, des vomissements importants et des diminutions rapides des glycémies. En cas de surdosage, un traitement symptomatique adéquat doit être initié en fonction des signes et des symptômes cliniques du patient.
Classe pharmacothérapeutique : Médicaments utilisés dans le diabète, analogues du Glucagon-like peptide-1 (GLP-1), code ATC : A10BJ01.
Mécanisme
d'action
L'exénatide est
un agoniste du récepteur du glucagon-like peptide-1 (GLP-1) présentant
plusieurs actions hypoglycémiantes du glucagon-like peptide-1 (GLP-1). La
séquence d'acides aminés de l'exénatide correspond partiellement à celle du
GLP-1 humain. In vitro, l'exénatide se lie et active les récepteurs
humains connus du GLP-1, son mécanisme d'action utilisant l'AMP cyclique et/ou
d'autres voies de transmission intracellulaires.
L'exénatide augmente de façon glucose-dépendante la sécrétion d'insuline par les cellules bêta pancréatiques. Lorsque la glycémie diminue, la sécrétion d'insuline diminue. Quand l'exénatide était associé à la metformine et/ou à une thiazolidinedione, aucune augmentation de l'incidence des hypoglycémies n'a été observée par rapport à la metformine et/ou une thiazolidinedione. Ceci peut être dû au mécanisme insulino-sécréteur glucose-dépendant (voir rubrique Mises en garde spéciales et précautions d'emploi).
L'exénatide inhibe la sécrétion de glucagon, connue pour être anormalement élevée dans le diabète de type 2. Des concentrations plus faibles de glucagon conduisent à une diminution de la production de glucose hépatique. En réponse à une hypoglycémie, l'exénatide n'inhibe cependant pas la réponse normale du glucagon et celle d'autres hormones.
L'exénatide ralentit la vidange gastrique diminuant ainsi le taux d'absorption intestinal du glucose.
Une diminution de la prise alimentaire due à une diminution de l'appétit et à une augmentation de la satiété a été montrée lors de l'administration d'exénatide.
Effets
pharmacodynamiques
L'exénatide améliore
le contrôle glycémique des patients ayant un diabète de type 2 en diminuant de
manière durable les glycémies à jeun et post-prandiales. Contrairement aux
GLP-1 endogènes, l'exénatide à libération prolongée a un profil
pharmacocinétique et pharmacodynamique approprié à une administration
hebdomadaire.
Une étude pharmacodynamique avec l'exénatide a démontré chez des patients ayant un diabète de type 2 (n=13) une restauration de la première phase de sécrétion d'insuline et une seconde phase de sécrétion d'insuline améliorée, en réponse à un bolus intraveineux de glucose.
Efficacité
et sécurité cliniques
Les résultats des
études cliniques à long terme de l'exénatide à libération prolongée sont
présentés ci- dessous, ces études ont inclus 1 356 sujets traités par
l'exénatide à libération prolongée, 52 % d'hommes et 48 % de femmes ; 230
sujets (17 %) étaient âgés de 65 ans et plus.
De plus, une étude de suivi cardiovasculaire, en double aveugle contre placebo (EXSCEL) a inclus 14 752 patients présentant un diabète de type 2 associé à un risque CV de tout niveau en ajout des traitements standards.
Contrôle
glycémique
Dans deux études,
l'exénatide à libération prolongée 2 mg hebdomadaire a été comparé à
l'exénatide 5 µg à libération immédiate administré deux fois par jour pendant 4
semaines suivi d'exénatide 10 µg à libération immédiate administré deux fois
par jour. Une des études durait 24 semaines (n= 252) et l'autre étude durait 30
semaines (n=295), suivies d'une période d'extension en ouvert au cours de laquelle
tous les patients étaient traités par l'exénatide à libération prolongée 2 mg
hebdomadaire pour une période supplémentaire de 7 ans (n=258). Dans ces études,
les baisses d'HbA1c étaient manifestes dans les deux groupes de traitement dès
la première mesure de l'HbA1c après traitement (semaines 4 ou 6).
L'exénatide à libération prolongée a conduit à une réduction statistiquement significative de l'HbA1c comparé aux patients recevant l'exénatide à libération immédiate (Tableau 2).
Un effet cliniquement pertinent sur l'HbA1c a été observé chez les patients traités par l'exénatide à libération prolongée et l'exénatide à libération immédiate, quel que soit le traitement antidiabétique de fond dans les deux études.
Dans les deux études, un nombre cliniquement et statistiquement significativement plus important de patients traités par l'exénatide à libération prolongée comparé à ceux traités par l'exénatide à libération immédiate a atteint une réduction de l'HbA1c ≤ 7 % ou < 7 % (p < 0,05 et p ≤ 0,0001 respectivement).
Les patients traités par l'exénatide à libération prolongée et ceux traités par l'exénatide à libération immédiate ont tous présenté une perte de poids par rapport au poids initial, mais la différence entre les deux bras de traitement n'était pas significative.
Dans
l'extension d'étude non contrôlée, les patients évaluables qui sont passés à la
semaine 30 de l'exénatide à libération immédiate à l'exénatide à libération
prolongée (n = 121), ont obtenu la même amélioration de l'HbA1c de -2,0% par
rapport à la valeur initiale à la semaine 52 que les patients traités avec
l'exénatide à libération prolongée.
Pour tous les patients qui ont terminé la phase d'extension d'étude non
contrôlée de 7 ans (n = 122 des 258 patients inclus dans la phase d'extension),
l'HbA1c a progressivement augmenté à partir de la semaine 52, mais elle était
toujours réduite après 7 ans par rapport à la valeur initiale (-1,5%). La perte
de poids a été maintenue pendant 7 ans chez ces patients.
Tableau 2 : Résultats de deux études cliniques avec l'exénatide à libération prolongée versus l'exénatide à libération immédiate associés à un régime alimentaire et une activité physique seuls, à la metformine et/ou un sulfamide hypoglycémiant et à la metformine et/ou une thiazolidinedione (patients en intention de traiter).
|
Etude de 24 semaines |
Exénatide à libération prolongée 2 mg |
Exénatide 10 µg à libération immédiate deux fois par jour |
|
N |
129 |
123 |
|
Taux d'HbA1c moyen (%) |
||
|
A l'inclusion |
8,5 |
8,4 |
|
Variation depuis l'inclusion (± SE) |
-1,6 (±0,1)** |
-0,9 (±0,1) |
|
Moyenne de la différence de variation depuis l'inclusion entre les traitements (95 % IC) |
-0,67 (-0,94 ; -0,39) ** |
|
|
Patients (%) ayant atteint une HbA1c < 7 % |
58 |
30 |
|
Variation de la glycémie à jeun (mmol/L) (± SE) |
-1,4 (±0,2) |
-0,3 (±0,2) |
|
Poids corporel moyen (kg) |
||
|
A l'inclusion |
97 |
94 |
|
Variation depuis l'inclusion (± SE) |
-2,3 (±0,4) |
-1,4 (± 0,4) |
|
Moyenne de la différence de variation depuis l'inclusion entre les traitements (95 % IC) |
-0,95 (-1,91 ; 0,01) |
|
|
Etude de 30 semaines |
||
|
N |
148 |
147 |
|
Taux d'HbA1c moyen (%) |
||
|
A l'inclusion |
8,3 |
8,3 |
|
Variation depuis l'inclusion (± SE) |
-1,9 (±0,1)* |
-1,5 (±0,1) |
|
Moyenne de la différence de variation depuis l'inclusion entre les traitements (95 % IC) |
-0,33 (-0,54 ; -0,12) * |
|
|
Patients (%) ayant atteint une HbA1c ≤ 7 % |
73 |
57 |
|
Variation de la glycémie à jeun (mmol/L) (± SE) |
-2,3 (±0,2) |
-1,4 (±0,2) |
|
Poids corporel moyen (kg) |
||
|
A l'inclusion |
102 |
102 |
|
Variation depuis l'inclusion (± SE) |
-3,7 (±0,5) |
-3,6 (±0,5) |
|
Moyenne de la différence de variation depuis l'inclusion entre les traitements (95 % IC) |
-0,08 (-1,29 ; 1,12) |
|
SE = erreur standard, IC= intervalle de confiance, * p< 0,05, **p< 0,0001
Une étude d'une durée de 26 semaines a été conduite au cours de laquelle l'exénatide à libération prolongée 2 mg a été comparé à l'insuline glargine une fois par jour. Par rapport au traitement par insuline glargine, l'exénatide à libération prolongée a démontré une variation de l'HbA1c supérieure, a diminué significativement le poids corporel moyen et était associé à moins d'épisodes hypoglycémiques (Tableau 3).
Tableau 3 : Résultats d'une étude de 26 semaines avec l'exénatide à libération prolongée versus insuline glargine en association avec la metformine avec ou sans sulfamide hypoglycémiant (patients en intention de traiter).
|
Exénatide à libération prolongée 2 mg |
Insuline glargine1 |
|
|
N |
233 |
223 |
|
Taux d'HbA1c moyen (%) |
||
|
A l'inclusion |
8,3 |
8,3 |
|
Variation depuis l'inclusion (± SE) |
-1,5 (± 0,1)* |
-1,3 (± 0,1)* |
|
Moyenne de la différence de variation depuis l'inclusion entre les traitements (95 % IC) |
-0,16 (-0,29 ; -0,03)* |
|
|
Patients (%) ayant atteint un taux d'HbA1c ≤ 7 % |
62 |
54 |
|
Variation de la glycémie à jeun (mmol/L) (± SE) |
-2,1 (± 0,2) |
-2,8 (± 0,2) |
|
Poids corporel moyen (kg) |
||
|
A l'inclusion |
91 |
91 |
|
Variation depuis l'inclusion (± SE) |
-2,6 (± 0,2) |
+1,4 (±0,2) |
|
Moyenne de la différence de variation depuis l'inclusion entre les traitements (95 % IC) |
-4,05 (-4,57 ; -3,52) * |
|
SE = erreur
standard, IC= intervalle de confiance, * p< 0,05
1La dose d'insuline glargine était titrée pour obtenir une glycémie
à jeun entre 4,0 et 5,5 mmol/L (72 à 100 mg/dL). La dose moyenne d'insuline
glargine en début de traitement était de 10,1 UI/jour et a augmenté jusqu'à
31,1 UI/jour pour les patients traités par insuline glargine.
Les résultats à 156 semaines étaient cohérents avec ceux rapportés précédemment dans le rapport intermédiaire à 26 semaines. Le traitement par l'exénatide à libération prolongée a permis une amélioration persistante et significative du contrôle glycémique et du contrôle du poids, comparativement au traitement par l'insuline glargine. Les résultats de tolérance à 156 semaines étaient cohérents avec ceux rapportés à 26 semaines.
Dans une étude de 26 semaines en double aveugle, l'exénatide à libération prolongée était comparé aux doses maximales quotidiennes de sitagliptine et de pioglitazone chez des sujets traités également par metformine. Tous les groupes de traitement présentaient une réduction significative de l'HbA1c comparé à la valeur initiale. L'exénatide à libération prolongée a démontré une supériorité par rapport à la fois à la sitagliptine et à la pioglitazone sur la variation de l'HbA1c par rapport à la valeur initiale.
L'exénatide à libération prolongée a démontré des réductions du poids significativement plus importantes comparé à la sitagliptine. Les patients traités par pioglitazone ont pris du poids (Tableau 4).
Tableau 4 : Résultats d'une étude de 26 semaines de l'exénatide à libération prolongée versus sitagliptine et versus pioglitazone en association à la metformine (patients en intention de traiter).
|
Exénatide à libération prolongée 2 mg |
Sitagliptine 100 mg |
Pioglitazone 45 mg |
|
|
N |
160 |
166 |
165 |
|
Taux d'HbA1c moyen (%) |
|||
|
A l'inclusion |
8,6 |
8,5 |
8,5 |
|
Variation depuis l'inclusion (± SE) |
-1,6 (± 0,1)* |
-0,9 (± 0,1)* |
-1,2 (± 0,1)* |
|
Moyenne de la différence de variation depuis l'inclusion entre les traitements (95 % IC) versus sitagliptine |
-0,63 (-0,89 ; -0,37)** |
||
|
Moyenne de la différence de variation depuis l'inclusion entre les traitements (95 % IC) versus pioglitazone |
-0,32 (-0,57 ; -0,06)* |
||
|
Patients (%) ayant atteint un taux d'HbA1c ≤ 7 % |
62 |
36 |
49 |
|
Variation de la glycémie à jeun (mmol/L) (± SE) |
-1,8 (± 0,2) |
-0,9 (± 0,2) |
-1,5 (± 0,2) |
|
Poids corporel moyen (kg) |
|||
|
A l'inclusion |
89 |
87 |
88 |
|
Variation depuis l'inclusion (± SE) |
-2,3 (± 0,3) |
-0,8 (± 0,3) |
+2,8 (± 0,3) |
|
Moyenne de la différence de variation depuis l'inclusion entre les traitements (95 % IC) versus sitagliptine |
-1,54 (-2,35 ; -0,72)* |
||
|
Moyenne de la différence de variation depuis l'inclusion entre les traitements (95 % IC) versus pioglitazone |
-5,10 (-5,91 ; -4,28)** |
||
SE = erreur standard, IC = intervalle de confiance, * p< 0,05, **p< 0,0001
Dans une étude de 28 semaines en double aveugle, l'association d'exénatide à libération prolongée et de dapagliflozine a été comparée à l'exénatide à libération prolongée seule et à la dapagliflozine seule chez des sujets traités également par de la metformine. Tous les groupes de traitement ont obtenu une diminution de l'HbA1C par rapport à la valeur initiale. Le groupe de traitement exénatide à libération prolongée et dapagliflozine a présenté des réductions supérieures de l'HbA1c par rapport à l'exénatide seule ou à la dapagliflozine seule en comparaison à la valeur initiale (Tableau 5).
L'association d'exénatide à libération prolongée et de dapagliflozine a démontré significativement une plus grande réduction du poids en comparaison à chaque médicament seul (Tableau 5).
Tableau 5 : Résultats d'une étude de 28 semaines comparant l'association d'exénatide à libération prolongée et de dapagliflozine à l'exénatide à libération prolongée seule et à la dapagliflozine seule, en association à la metformine (patients en intention de traiter).
|
Exénatide à libération prolongée 2 mg QW + Dapagliflozine 10 mg QD |
Exénatide à libération prolongée 2 mg QW + Placebo QD |
Dapagliflozine 10 mg QD + Placebo QW |
|
|
N |
228 |
227 |
230 |
|
Taux d'HbA1c moyen (%) |
|||
|
A l'inclusion |
9,3 |
9,3 |
9,3 |
|
Variation depuis l'inclusion (± SE)a |
-2,0 (±0,1) |
-1,6 (±0,1) |
-1,4 (±0,1) |
|
Moyenne de la différence de variation depuis l'inclusion entre l'association et le médicament actif seul (95 % IC) |
-0,38* (-0,63; -0,13) |
-0,59** (-0,84; -0,34) |
|
|
Patients (%) ayant atteint un taux d'HbA1c < 7 % |
45 |
27 |
19 |
|
Moyenne de la variation depuis l'inclusion de la glycémie à jeun (mmol/L) (±SE)a |
-3,7 (±0,2) |
-2,5 (±0,2) |
-2,7 (±0,2) |
|
Moyenne de la différence de variation depuis l'inclusion entre l'association et le médicament actif seul (95 % IC) |
-1,12** (-1,55 ; -0,68) |
-0,92** (-1,36 ; -0,49) |
|
|
Moyenne de la variation depuis l'inclusion de la glycémie postprandiale à 2 heures (mmol/L) (?SE)a |
-4,9 (±0,2) |
-3,3 (±0,2) |
-3,4 (±0,2) |
|
Moyenne de la différence de variation depuis l'inclusion entre l'association et le médicament actif seul (95 % IC) |
-1,54** (-2,10 ; -0,98) |
-1,49** (-2,04 ; -0,93) |
|
|
Poids corporel moyen (kg) |
|||
|
A l'inclusion |
92 |
89 |
91 |
|
Variation depuis l'inclusion (± SE)a |
-3,6 (±0,3) |
-1,6 (±0,3) |
-2,2 (±0,3) |
|
Moyenne de la différence de variation depuis l'inclusion entre l'association et le médicament actif seul (95 % IC) |
-2,00** (-2,79; -1,20) |
-1,33** (-2,12; -0,55) |
|
QW= une fois
par semaine, QD= une fois par jour, SE = erreur standard, CI= intervalle de
confiance, N=nombre de patients.
a La moyenne ajustée des moindres carrés et la différence entre les
différents groupes de traitement de la variation entre les valeurs à
l'inclusion et la semaine 28 ont été modellisées en utilisant un modèle mixte à
mesures répétées (MMMR) incluant comme facteurs fixes le traitement, la région,
la catégorie de l'HbA1C à l'inclusion (< 9,0 % or ≥ 9,0 %), la
semaine, et le traitement par semaine et la valeur à l'inclusion en tant que
co-variante.
*p < 0.01, **p < 0.001.
Les valeurs de p sont toutes des valeurs de p ajustées pour des mesures
multiples.
Les analyses excluent les mesures post traitement et après arrêt prématuré de
la prise des produits à l'étude.
Dans une étude
en double aveugle de 28 semaines, l'exénatide à libération prolongée ajouté à
l'insuline glargine seule ou avec la metformine a été comparé au placebo ajouté
à l'insuline glargine seule ou avec la metformine. L'insuline glargine ciblant
une glycémie à jeun entre 4,0 et 5,5 mmol/L (72 à 99 mg/dL) a été titrée.
L'exénatide à libération prolongée a démontré une supériorité au placebo en
diminuant l'HbA1c de la valeur initiale à la semaine 28 (Tableau 6).
Pour la diminution du poids corporel, l'exénatide à libération prolongée a été
supérieur au placebo à la semaine 28 (Tableau 6).
Tableau 6 : Résultats d'une étude de 28 semaines de l'exénatide à libération prolongée versus placebo en association avec l'insuline glargine seule ou avec la metformine (patients en intention de traiter)
|
Exénatide à libération prolongée 2 mg + Insuline glarginea |
Placebo + Insuline glarginea |
|
|
N |
230 |
228 |
|
Taux d'HbA1c moyen (%) |
||
|
A l'inclusion |
8,5 |
8,5 |
|
Variation depuis l'inclusion (± SE)b |
-1,0 (±0,1) |
-0,2 (±0,1) |
|
Moyenne de la différence de variation depuis |
-0,74* |
|
|
l'inclusion entre les traitements (95 % IC) |
(-0,94 ; -0,54) |
|
|
Patients (%) ayant atteint un taux d'HbA1c ≤7 %c |
33* |
7 |
|
Poids corporel moyen (kg) |
||
|
A l'inclusion |
94 |
94 |
|
Variation depuis l'inclusion (± SE)b |
-1,0 (±0,3) |
0,5 (±0,3) |
|
Moyenne de la différence de variation depuis |
-1,52* |
|
|
l'inclusion entre les traitements (95 % IC) |
(-2,19 ; -0,85) |
|
|
Variation de la glycémie postprandiale à 2 heures depuis l'inclusion (mmol/L) (?SE)b,d |
-1,6 (±0,3) |
-0,1 (±0,3) |
|
Moyenne de la différence de variation depuis |
-1,54* |
|
|
l'inclusion entre les traitements (95 % IC) |
(-2,17 ; -0,91) |
|
a. La variation de la moyenne des moindres carrés de la dose journalière moyenne était de 1,6 unités pour le groupe de l'exénatide à libération prolongée et 3,5 unités pour le groupe placebo.
b. La moyenne ajustée des moindres carrés et la différence entre les différents groupes de traitement de la variation entre les valeurs à l'inclusion et la semaine 28 ont été modellisées en utilisant un modèle mixte à mesures répétées (MMMR) incluant comme facteurs fixes le traitement, la région, la catégorie de l'HbA1C à l'inclusion (< 9,0 % or ≥ 9,0 %), l'utilisation de SU (oui, non), la semaine, et le traitement par semaine et la valeur à l'inclusion en tant que co-variante. La variation absolue de la glycémie post-prondiale à 2 heures à la semaine 28 est modellisée de manière similaire en utilisant ANCOVA.
c. Tous les patients avec des données manquantes ont été considérés comme non répondeurs.
d. Après un repas test.
Les analyses excluent les mesures d'urgence post traitement ainsi que l'arrêt prématuré de la prise des médicaments de l'étude.
Évaluation
cardiovasculaire
EXSCEL était
une étude pragmatique de suivi cardiovasculaire (CV) chez les patients atteints
de diabète de type 2 avec un risque CV de tout niveau. Un total de 14 752
patients ont été randomisés 1:1, soit à l'exénatide à libération prolongée 2 mg
une fois par semaine soit au placebo, ajouté aux soins standards actuels qui
pouvaient inclure des inhibiteurs de SGLT2. Les patients ont été suivis comme
dans la pratique clinique standard pendant une période médiane de 38,7 mois
avec une durée médiane de traitement de 27,8 mois. Le statut vital était connu
à la fin de l'étude pour 98,9 % et 98,8 % des patients du groupe exénatide à
libération prolongée et du groupe placebo, respectivement. L'âge moyen à
l'entrée dans l'étude était de 62 ans (avec 8,5 % des patients âgés de 75 ans
et plus). Environ 62 % des patients étaient des hommes. L'IMC moyen était de
32,7 kg / m2 et la durée moyenne du diabète était de 13,1 ans. Le taux moyen
d'HbA1c était de 8,1 %. Environ 49,3 % présentaient une insuffisance rénale
légère (débit de filtration glomérulaire estimé [DFGe] ≥60 à ≤ 89
mL / min / 1,73 m2) et 21,6 % présentaient une insuffisance rénale modérée (DFG
≥ 30 à ≤ 59 mL / min / 1,73 m2). Dans l'ensemble, 26,9 % des
patients n'avaient jamais eu d'événement CV, 73,1 % avaient au moins un
événement CV antérieur.
Le critère principal de sécurité (non infériorité) et d'efficacité (supériorité) dans EXSCEL était le délai de survenue du premier événement cardiaque majeur (MACE) confirmé : décès cardiovasculaire, infarctus du myocarde non fatal ou accident vasculaire cérébral non fatal. La mortalité toutes causes confondues était le critère d'évaluation secondaire initial évalué.
L'exénatide à libération prolongée n'a pas augmenté le risque cardiovasculaire chez les patients atteints de diabète de type 2 par rapport au placebo lorsqu'il a été ajouté aux soins standards actuels (HR : 0,91; IC à 95 %: 0,832 ; 1,004 p < 0,001 pour une non infériorité), voir figure 1. Dans l'analyse pré- spécifiée d'un sous-groupe de l'étude EXSCEL, le HR du MACE était de 0,86 (IC à 95% : 0,77 - 0,97) chez des patients avec un débit de filtration glomérulaire estimé à l'inclusion [DFGe] ≥ 60 mL/min/1,73 m2 et de 1,01 (IC à 95% : 0,86 - 1,19) chez des patients avec un DFGe à l'inclusion < 60 mL/min/1,73 m2. Les résultats cardiovasculaires des critères composites primaires et secondaires sont présentés à la figure 2.
Figure 1 : Temps jusqu'au premier MACE (en intention de traiter les patients)
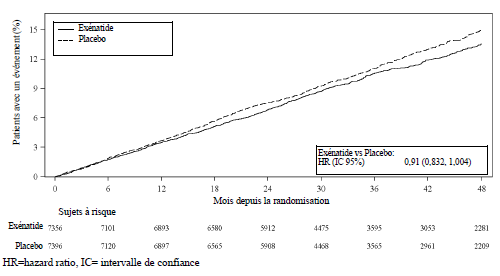
Figure 2 : Forest Plot : Analyse des critères d'évaluation primaires et secondaires (en intention de traiter les patients)
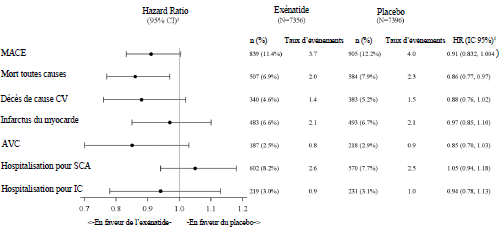
SCA = syndrome
coronaire aigu ; IC = intervalle de confiance ; CV = cardiovasculaire ; IC =
insuffisance cardiaque ; HR = hazard ratio; MACE = événement cardiaque
indésirable majeur; IM = infarctus du myocarde; n = nombre de patients avec un
événement; N = nombre de patients dans le groupe de traitement.
1 Le HR (actif
/ placebo) et l'IC sont basés sur le modèle de régression des risques
proportionnels de Cox, stratifiés par événement CV antérieur, avec le groupe de
traitement uniquement comme variable explicative.
La nécessité d'un traitement antihyperglycémiant supplémentaire a été diminué de 33% dans le groupe exénatide à libération prolongée (incidence ajustée en fonction de l'exposition de 10,5 pour 100 patients-année) par rapport au groupe placebo (incidence corrigée de l'exposition de 15,7 pour 100 patients-année). Une réduction du taux d'HbA1c a été observée au cours de l'ensemble de l'essai avec une différence de traitement de -0,53 % (exénatide à libération prolongée par rapport au placebo).
Poids
corporel
Une réduction du
poids corporel comparé au poids à l'inclusion a été observée dans toutes les
études avec l'exénatide à libération prolongée. Dans les 4 études contrôlées
avec comparateurs, cette réduction du poids a été observée chez les patients
traités par l'exénatide à libération prolongée indépendamment de la présence de
nausées, bien que la réduction fût plus importante dans le groupe ayant des
nausées (réduction moyenne de - 2,9 kg à -5,2 kg pour les patients ayant des
nausées versus de - 2,2 kg à -2,9 kg pour ceux n'ayant pas de nausées).
Dans les 4 études contrôlées avec comparateurs, le pourcentage de patients ayant présenté une perte de poids associée à une diminution de l'HbA1c est compris entre 70 et 79 % (le pourcentage de patients ayant présenté une diminution de l'HbA1c est compris entre 88 et 96 %).
Glucose
plasmatique/sérique
Le traitement
avec l'exénatide à libération prolongée a conduit à des diminutions
significatives de la glycémie plasmatique/sérique à jeun, ces diminutions ont
été observées dès 4 semaines. Dans l'étude contrôlée versus placebo avec
l'insuline glargine, la variation de la glycémie à jeun depuis l'inclusion
jusqu'à la semaine 28 était de - 0,7 mmol/L pour le groupe de l'exénatide à
libération prolongée et de - 0,1 mmol/L pour le groupe placebo. De plus, des
diminutions des concentrations postprandiales ont également été observées.
L'amélioration des glycémies à jeun était durable pendant 52 semaines.
Fonction
bêta-cellulaire
Les études
cliniques avec l'exénatide à libération prolongée mettent en évidence une
amélioration de la fonction bêta-cellulaire, en utilisant des mesures telles
que le modèle d'homéostasie (HOMA-B). La pérennité de l'effet sur la fonction
bêta-cellulaire était maintenue durant 52 semaines.
Pression
artérielle
Une diminution
de la pression artérielle systolique a été observée au cours des 4 études
contrôlées avec l'exénatide à libération prolongée (de 2,9 mmHg à 4,7 mmHg).
Dans l'étude comparative avec l'exénatide à libération immédiate sur 30
semaines, l'exénatide à libération prolongée et l'exénatide à libération
immédiate ont diminué significativement la pression artérielle systolique par
rapport à la valeur initiale (4,7±1,1 mmHg et 3,4±1,1 mmHg respectivement), la
différence entre les groupes de traitement n'était pas significative. Des
améliorations de la pression artérielle étaient maintenues sur 52 semaines.
Dans l'étude contrôlée versus placebo avec l'insuline glargine la variation de la pression artérielle systolique depuis l'inclusion jusqu'à la semaine 28 était de - 2,6 mmHg pour le groupe de l'exénatide à libération prolongée et de - 0,7 mmHg pour le groupe placebo.
Le traitement avec l'association d'exénatide à libération prolongée et de dapagliflozine a conduit à la semaine 28 à une diminution moyenne significative de -4,3±0,8 mmHg de la pression artérielle systolique comparée à celle avec l'exénatide à libération prolongée seule de -1,2±0,8 mmHg (p < 0,01) ou à celle avec la dapagliflozine seule de -1,8±0,8 mmHg (p < 0,05).
Lipides à
jeun
L'exénatide à
libération prolongée n'a pas montré d'effets négatifs sur les paramètres
lipidiques.
Population
pédiatrique
L'Agence
Européenne du Médicament a différé l'obligation de soumettre les résultats
d'études réalisées avec l'exénatide à libération prolongée dans un ou plusieurs
sous-groupes de la population pédiatrique dans le diabète de type 2 (voir
rubrique Posologie et mode d'administration pour les informations
concernant l'usage pédiatrique).
Les propriétés d'absorption de l'exénatide reflètent les propriétés de libération prolongée de la formulation de l'exénatide à libération prolongée. Une fois absorbé dans la circulation, exénatide est distribué et éliminé conformément à ses propriétés systémiques pharmacocinétiques connues (tel que décrit dans cette rubrique).
Absorption
Après
administration hebdomadaire de 2 mg de l'exénatide à libération prolongée, les
concentrations moyennes de l'exénatide dépassaient les concentrations moyennes
efficaces (~ 50 pg/mL) en 2 semaines avec une augmentation graduelle de la
concentration plasmatique moyenne d'exénatide pendant plus de 6 à 7 semaines.
Par la suite, des concentrations d'exénatide d'approximativement 151-265 pg/mL
ont été maintenues, indiquant que l'état d'équilibre était atteint. Les
concentrations d'exénatide à l'état d'équilibre étaient maintenues pendant
l'intervalle d'une semaine entre les doses avec une fluctuation minimale en pic
et en creux à partir de cette concentration thérapeutique moyenne.
Distribution
Après
administration sous-cutanée d'une dose unique d'exénatide, le volume de
distribution apparent moyen de l'exénatide est de 28 L.
Biotransformation
et élimination
Des études
précliniques ont montré que l'exénatide est principalement éliminé par
filtration glomérulaire, suivie d'une dégradation protéolytique. La clairance
apparente moyenne de l'exénatide est de 9 L/h. Ces caractéristiques
pharmacocinétiques de l'exénatide sont indépendantes de la dose.
Approximativement 10 semaines après l'arrêt du traitement par l'exénatide à
libération prolongée, les concentrations moyennes plasmatiques d'exénatide ont
diminué sous des concentrations minimales détectables.
Populations particulières
Atteinte de
la fonction rénale
Une analyse
pharmacocinétique d'une population de patients ayant une insuffisance rénale
recevant l'exénatide à libération prolongée 2 mg, indique qu'il peut y avoir
une augmentation de l'exposition d'approximativement 74 % et 23 % (prédiction
médiane dans chaque groupe) respectivement chez les insuffisants rénaux modérés
(N=10) et légers (N=56), comparé aux patients ayant une fonction rénale normale
(N=84).
Atteinte de la fonction hépatique
Aucune
étude pharmacocinétique n'a été effectuée chez les patients ayant une
insuffisance hépatique. L'exénatide étant principalement éliminée par le rein,
l'insuffisance hépatique ne devrait pas modifier les concentrations sanguines
de l'exénatide.
Sexe, race et poids
Le sexe, la
race et le poids n'ont aucune influence cliniquement significative sur la
pharmacocinétique de l'exénatide.
Sujets âgés
Les données
chez les patients âgés sont limitées, mais suggèrent que jusqu'à environ 75
ans, il n'y a pas de modifications importantes de l'exposition à l'exénatide.
Dans une étude de pharmacocinétique avec l'exénatide à libération immédiate chez des patients ayant un diabète de type 2, l'administration d'exénatide (10 µg) a entraîné une augmentation moyenne de l'ASC de l'exénatide de 36 % chez 15 sujets âgés de 75 à 85 ans, par rapport à 15 sujets de 45 à 65 ans. Cette augmentation est susceptible d'être liée à une fonction rénale diminuée dans le groupe le plus âgé (voir rubrique Posologie et mode d'administration).
Population
pédiatrique
Lors d'une
étude de pharmacocinétique en dose unique de l'exénatide à libération immédiate
réalisée chez 13 patients avec un diabète de type 2 âgés de 12 à 16 ans,
l'administration de l'exénatide (5 µg) s'est traduite par une ASC et une Cmax
légèrement plus basses (de 16 % pour l'ASC et de 25 % pour la Cmax) que celles
observées chez l'adulte. Aucune étude de pharmacocinétique avec l'exénatide à
libération prolongée n'a été conduite dans la population pédiatrique.
L'exénatide à libération prolongée a une influence mineure sur l'aptitude à conduire des véhicules et à utiliser des machines. Lorsque l'exénatide à libération prolongée est utilisé en association avec un sulfamide hypoglycémiant, les patients doivent être informés des précautions à prendre afin d'éviter une hypoglycémie lors de la conduite de véhicules et l'utilisation de machines.
Les données non cliniques issues des études conventionnelles de pharmacologie de sécurité, toxicologie en administration répétée, ou génotoxicité conduites avec l'exénatide à libération immédiate ou l'exénatide à libération prolongée n'ont pas révélé de risque particulier pour l'homme.
Des tumeurs thyroïdiennes ont été observées chez le rat et la souris ayant reçu des agonistes du récepteur du GLP-1 à longue durée d'action. Dans une étude de carcinogénicité de 2 ans conduite chez le rat avec l'exénatide à libération prolongée, une augmentation de l'incidence des adénomes et des carcinomes à cellules C a été observée à des doses ≥ 2 fois supérieures à l'exposition systémique humaine basée sur l'ASC. La pertinence clinique de ces résultats n'est actuellement pas connue.
Les études chez l'animal avec l'exénatide n'ont pas montré d'effet délétère sur la fertilité ; des doses élevées d'exénatide ont entrainé des effets sur le squelette ainsi qu'une diminution de la croissance fœtale et néonatale.
Les stylos préremplis sont exclusivement à usage unique.
Le stylo doit être sorti du réfrigérateur au moins 15 minutes avant l'injection. La poudre contenue dans l'une des chambres doit être mélangée avec le solvant contenu dans l'autre chambre du stylo prérempli. Le solvant doit être inspecté visuellement avant l'utilisation. Le solvant doit être utilisé uniquement s'il est limpide et sans particule. Après mise en suspension, le mélange doit être utilisé uniquement s'il est blanc à blanc cassé et trouble. Veuillez-vous reporter à la notice et aux instructions d'utilisation pour des informations complémentaires sur la suspension et l'administration.
Utiliser uniquement les aiguilles spécifiques fournies avec le stylo.
L'exénatide à libération prolongée doit être injecté par voie sous-cutanée immédiatement après le mélange de la poudre et du solvant.
L'exénatide à libération prolongée ne doit pas être utilisé s'il a été congelé.
Il doit être indiqué au patient d'éliminer le stylo en respectant les règles de sécurité, l'aiguille encore fixée, après chaque injection.
Tout produit non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
liste I
Remboursement en fonction de l'indication : (JO du 12/05/2015)
Les seules indications thérapeutiques ouvrant droit à la
prise en charge ou au remboursement par l'assurance maladie sont
:
- Traitement du diabète de type 2 en
association :
. à la metformine ;
. aux sulfamides hypoglycémiants ;
. à la metformine et un sulfamide
hypoglycémiant.
- Chez les adultes n'ayant pas obtenu un contrôle glycémique adéquat aux doses maximales tolérées de ces traitements oraux.
Poudre et solvant pour suspension injectable à libération prolongée.
Poudre : de couleur blanche à blanc cassé.
Solvant : solution claire, d'incolore à jaune pâle ou brun pâle.
Chaque
stylo à double chambre contient de la poudre d'exénatide et du solvant
dans une cartouche de verre de type 1, scellée à une extrémité par un
bouchon de caoutchouc chlorobutyl et un opercule en aluminium, et à
l'autre extrémité par un piston en caoutchouc chlorobutyl. Les deux
chambres sont séparées par un deuxième piston en caoutchouc chlorobutyl.
Chaque stylo est muni d'une aiguille. Chaque boîte contient également
une aiguille de rechange. Utiliser uniquement les aiguilles fournies
avec le stylo.
Conditionnement de 4 stylos préremplis à dose unique.
Chaque stylo pré-rempli contient 2 mg d'exénatide. Après mise en suspension, chaque stylo délivre une dose de 2 mg dans 0,65 mL.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Poudre
poly (D,L-lactide-co-glycolide)
saccharose
Solvant
carmellose sodique
chlorure de sodium
polysorbate 20
dihydrogéno-phosphate de sodium, monohydraté
phosphate de disodium, heptahydraté
eau pour injection
hydroxyde de sodium (pour l'ajustement du pH)