
XTANDI 40 mg, capsule molle, boîte de 4 plaquettes thermoformées de 28
Retiré du marché le : 13/05/2019
Dernière révision : 23/10/2018
Taux de TVA : 2.1%
Prix de vente : 2 821,58 €
Taux remboursement SS : 100%
Base remboursement SS : 2 821,58 €
Laboratoire exploitant : ASTELLAS PHARMA FRANCE
Source :

Xtandi est indiqué dans :
le traitement du cancer de la prostate résistant à la castration (CPRC) non métastatique à haut risque chez les hommes adultes (voir rubrique Propriétés pharmacodynamiques).
le traitement du CPRC métastatique chez les hommes adultes asymptomatiques ou peu symptomatiques, après échec d'un traitement par suppression androgénique et pour lesquels la chimiothérapie n'est pas encore cliniquement indiquée (voir rubrique Propriétés pharmacodynamiques).
le traitement du CPRC métastatique chez les hommes adultes dont la maladie a progressé pendant ou après une chimiothérapie à base de docétaxel.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Composition. Femmes enceintes ou susceptibles de l'être (voir rubriques Grossesse et allaitement et Instructions pour l'utilisation, la manipulation et l'élimination).
Risque de convulsions
L'utilisation de l'enzalutamide a été associée à des convulsions (voir rubrique Effets indésirables). La décision de poursuivre le traitement chez les patients qui présentent des convulsions doit être évaluée au cas par cas.
Syndrome d'encéphalopathie postérieure réversible
De rares cas de Syndrome d'Encéphalopathie Postérieure Réversible (SEPR) ont été rapportés chez des patients traités par Xtandi (voir rubrique Effets indésirables). Le SEPR est un trouble neurologique rare, réversible, pouvant se manifester par la survenue rapide des symptômes suivants : convulsions, céphalées, confusion, cécité et autres troubles de la vision ou troubles neurologiques, avec ou sans hypertension associée. Le diagnostic de SEPR requiert une confirmation par imagerie cérébrale, de préférence par imagerie par résonance magnétique (IRM). Chez les patients qui développent un SEPR, l'arrêt du traitement par Xtandi est recommandé.
Utilisation concomitante d'autres médicaments
L'enzalutamide est un inducteur enzymatique puissant et peut entraîner une diminution de l'efficacité de nombreux médicaments couramment utilisés (voir les exemples en rubrique Interactions avec d'autres médicaments et autres formes d'interactions). Une réévaluation des traitements concomitants doit être conduite à l'initiation du traitement par l'enzalutamide. L'utilisation concomitante de l'enzalutamide et de médicaments qui sont des substrats cibles de nombreuses enzymes du métabolisme ou de transporteurs (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions) doit généralement être évitée si leurs effets thérapeutiques sur le patient sont importants et si leur posologie ne peut pas être facilement ajustable sur la base du suivi de l'efficacité ou des concentrations plasmatiques.
L'administration concomitante de warfarine ou d'anticoagulants coumariniques doit être évitée. Si Xtandi est administré en même temps qu'un anticoagulant métabolisé par le CYP2C9 (tel que la warfarine ou l'acénocoumarol), une surveillance additionnelle du rapport normalisé international (INR) doit être conduite (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Insuffisance rénale
La prudence est recommandée en cas d'utilisation chez des patients présentant une insuffisance rénale sévère, l'enzalutamide n'ayant pas été étudié dans cette population de patients.
Insuffisance hépatique sévère
Un allongement de la demi-vie de l'enzalutamide a été observé chez les patients présentant une insuffisance hépatique sévère, peut-être lié à une augmentation de la distribution tissulaire. La pertinence clinique de cette observation reste inconnue. Un allongement du temps nécessaire pour atteindre l'état d'équilibre des concentrations est toutefois prévisible ; de même, il pourrait être constaté un allongement du temps nécessaire pour atteindre l'effet pharmacologique maximal ainsi que du temps jusqu'à l'apparition et jusqu'au déclin de l'induction enzymatique (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Antécédents récents de maladies cardiovasculaires
Les patients présentant des antécédents récents d'infarctus du myocarde (au cours des 6 mois précédents) ou d'angor instable (au cours des 3 mois précédents), une insuffisance cardiaque de classe III ou IV selon la classification de la New York Heart Association (NYHA) sauf en cas de fraction d'éjection ventriculaire gauche (FEVG) supérieure ou égale à 45 %, une bradycardie ou une hypertension non contrôlée ont été exclus des études de phase III. Il convient d'en tenir compte lorsque Xtandi est prescrit à des patients présentant ces caractéristiques.
Un traitement par suppression androgénique peut allonger l'intervalle QT
Chez les patients présentant des antécédents ou des facteurs de risques de l'allongement de l'intervalle QT, et chez les patients recevant de manière concomitante des médicaments susceptibles d'allonger l'intervalle QT (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions), les médecins doivent évaluer le rapport bénéfice / risque en prenant en compte le risque potentiel de torsades de pointes avant l'initiation du traitement par Xtandi.
Chimiothérapie concomitante
La sécurité d'emploi et l'efficacité de l'utilisation concomitante de Xtandi et d'une chimiothérapie cytotoxique n'ont pas été établies. L'administration concomitante d'enzalutamide n'a pas d'effet cliniquement significatif sur la pharmacocinétique du docétaxel administré par voie intraveineuse (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions) ; cependant, une hausse de la fréquence de neutropénie induite par le docétaxel ne peut être exclue.
Excipients
Xtandi contient du sorbitol (E420). Les patients présentant une intolérance héréditaire rare au fructose ne doivent pas prendre ce médicament.
Réactions d'hypersensibilité
Des réactions d'hypersensibilité ont été observées avec l'enzalutamide, se manifestant par des symptômes incluant, mais pas uniquement, un rash, ou un œdème du visage, de la langue, des lèvres ou du pharynx (voir rubrique Effets indésirables).
Résumé du profil de sécurité
Les effets indésirables les plus fréquents sont l'asthénie/fatigue, les bouffées de chaleur, les fractures et l'hypertension. Les autres effets indésirables importants comprennent la chute, les troubles cognitifs et la neutropénie.
Des cas de convulsions ont été rapportés chez 0,4 % des patients traités par enzalutamide, chez 0,1 % des patients sous placebo et chez 0,3 % des patients traités par bicalutamide.
De rares cas de Syndrome d'Encéphalopathie Postérieure Réversible ont été rapportés chez des patients traités par enzalutamide (voir rubrique Mises en garde et précautions d'emploi).
Liste tabulée des effets indésirables
Les effets indésirables observés au cours des études cliniques sont listés ci-dessous par catégorie de fréquence. Les catégories sont définies comme suit : très fréquent (≥ 1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, < 1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Les effets indésirables sont classés par ordre de gravité décroissant dans chaque catégorie de fréquence.
Tableau 1 : Effets indésirables observés au cours des études cliniques comparatives et post- commercialisation
| Classes de systèmes d'organes selon MedDRA | Effet indésirable et fréquence |
| Affections hématologiques et du système lymphatique | Peu fréquent : leucopénie, neutropénie Fréquence indéterminée * : thrombopénie |
| Affections du système immunitaire | Fréquence indéterminée * : œdème du visage, œdème de la langue, œdème labial, œdème pharyngé |
| Affections psychiatriques | Fréquent : anxiété Peu fréquent : hallucination visuelle |
| Affections du système nerveux | Fréquent : céphalées, trouble de la mémoire, amnésie, troubles de l'attention, syndrome des jambes sans repos Peu fréquent : troubles cognitifs, convulsions¥Fréquence indéterminée * : syndrome d'encéphalopathie postérieure réversible |
| Affections cardiaques | Fréquent : cardiopathie ischémique† Fréquence indéterminée * : allongement de l'intervalle QT (voir rubriques Mises en garde et précautions d'emploi et Interactions avec d'autres médicaments et autres formes d'interactions) |
| Affections vasculaires | Très fréquent : bouffées de chaleur, hypertension |
| Affections gastro-intestinales | Fréquence indéterminée * : nausées, vomissements, diarrhée |
| Affections de la peau et du tissu sous-cutané | Fréquent : sécheresse cutanée, prurit Fréquence indéterminée * : rash |
| Affections musculo-squelettiques et systémiques | Très fréquent : fractures‡ Fréquence indéterminée * : myalgie, spasmes musculaires, faiblesse musculaire, dorsalgie |
| Affections des organes de reproduction et du sein | Fréquent : gynécomastie |
| Troubles généraux et anomalies au site d'administration | Très fréquent : asthénie, fatigue |
| Lésions, intoxications et complications liées aux procédures | Fréquent : chute |
* Notifications spontanées issues de l'expérience post-commercialisation
¥ Évalué par la requête standardisée du dictionnaire MedDRA (SMQ) étroite de « Convulsions », incluant convulsion, convulsion grand mal, crises partielles complexes, crises partielles et état de mal épileptique. Cela inclut les rares cas de convulsions avec des complications mortelles.
† Évalué par les SMQ étroites de « Infarctus du myocarde » et « Autres cardiopathies ischémiques » incluant les termes préférés suivants, observés chez au moins deux patients dans les études de phase III randomisées, contrôlées versus placebo : angor, maladie coronarienne, infarctus du myocarde, infarctus du myocarde aigu, syndrome coronarien aigu, angor instable, ischémie myocardique et artériosclérose coronaire.
‡ Inclut tous les termes préférés comportant le mot « fracture » osseuse.
Description d'une sélection d'effets indésirables
Convulsions
Dans les études cliniques comparatives, 13 (0,4 %) des 3179 patients traités à la dose quotidienne de 160 mg d'enzalutamide ont présenté des convulsions, alors que un patient (0,1 %) parmi ceux ayant reçu le placebo et un patient (0,3 %) parmi ceux ayant reçu du bicalutamide ont présenté des convulsions. La dose semble être un facteur prédictif important du risque de convulsions, comme l'indiquent des données précliniques et les données obtenues lors d'une étude de recherche de dose. Dans les deux études cliniques comparatives, les patients présentant des antécédents de convulsions ou des facteurs de risque de convulsions ont été exclus.
Dans l'essai simple-bras 9785-CL-0403 (UPWARD) évaluant l'incidence des convulsions chez les patients présentant des facteurs de prédisposition aux convulsions (dont 1,6% avaient des antécédents de convulsions), 8 (2,2%) des 366 patients traités par enzalutamide ont présenté des convulsions. La durée moyenne de traitement était de 9,3 mois.
Le mécanisme par lequel l'enzalutamide pourrait abaisser le seuil épileptogène est inconnu, mais pourrait être mis en rapport avec les données des études in vitro qui montrent que l'enzalutamide et son métabolite actif se lient au canal chlore du récepteur GABA et peuvent en inhiber l'activité.
Cardiopathie ischémique
Dans les études cliniques randomisées, contrôlées versus placebo, une cardiopathie ischémique est survenue chez 2,5 % des patients recevant l'enzalutamide plus un traitement par suppression androgénique versus
1,3 % des patients recevant le placebo plus un traitement par suppression androgénique.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
ARRETER LE TRAITEMENT ET CONSULTER UN MEDECIN DES QUE POSSIBLE en cas de :
- convulsions
- maux de tête
- confusion
- cécité ou autres troubles de la vision.
PREVENIR LE MEDECIN en cas de:
- éruption cutanée
- gonflement du visage, de la langue, des lèvres ou de la gorge.
PRUDENCE en cas d'utilisation de machine(s) ou de conduite de véhicule(s) (convulsions).
CONTRACEPTION :
Un préservatif doit être utilisé pendant la durée du traitement ainsi
que durant les 3 mois suivant son arrêt en cas de rapports sexuels avec
une femme enceinte ou en âge de procréer.
Femmes en âge de procréer
Aucune donnée n'est disponible sur l'utilisation de Xtandi chez la femme enceinte, et ce médicament ne doit pas être utilisé chez la femme en âge de procréer. Il se peut que ce médicament soit nocif pour l'enfant à naître ou entraîne un avortement spontané s'il est pris pendant la grossesse (voir rubriques Contre-indications, Données de sécurité précliniques et Instructions pour l'utilisation, la manipulation et l'élimination).
Contraception chez les hommes et les femmes
La présence de l'enzalutamide ou de ses métabolites dans le sperme n'est pas connue. L'utilisation d'un préservatif est nécessaire pendant le traitement et durant les trois mois suivant la fin du traitement par enzalutamide en cas de rapport sexuel avec une femme enceinte. En cas de rapport sexuel avec une femme en âge de procréer, l'utilisation d'un préservatif associé à une autre méthode de contraception efficace est nécessaire pendant le traitement et durant les trois mois suivant la fin du traitement. Des études menées chez l'animal ont fait état d'une toxicité sur la reproduction (voir rubrique Données de sécurité précliniques).
Grossesse
L'enzalutamide ne doit pas être utilisé chez la femme. L'enzalutamide est contre-indiqué chez la femme enceinte ou susceptible de l'être (voir rubriques Contre-indications, Données de sécurité précliniques et Instructions pour l'utilisation, la manipulation et l'élimination).
Allaitement
L'enzalutamide ne doit pas être utilisé chez la femme. On ne sait pas si l'enzalutamide est retrouvé dans le lait maternel humain. L'enzalutamide et/ou ses métabolites sont sécrétés dans le lait de rate (voir rubrique Données de sécurité précliniques).
Fertilité
Les études menées chez l'animal ont montré que l'enzalutamide affectait le système reproducteur des rats et des chiens mâles (voir rubrique Données de sécurité précliniques).
Effet potentiel d'autres médicaments sur l'exposition à l'enzalutamide
Inhibiteurs du CYP2C8
Le CYP2C8 joue un rôle important dans l'élimination de l'enzalutamide et dans la formation de son métabolite actif. Après administration par voie orale de gemfibrozil (600 mg deux fois par jour), un inhibiteur puissant du CYP2C8, chez des sujets sains de sexe masculin, l'ASC de l'enzalutamide a été augmentée d'environ 326 % tandis que sa Cmax a été diminuée de 18 %. L'ASC de la somme des fractions libres de l'enzalutamide et de son métabolite actif, a été augmentée de 77 % tandis que la Cmax a été diminuée de 19 %. Les inhibiteurs puissants (ex : gemfibrozil) du CYP2C8 doivent être évités ou utilisés avec précaution pendant le traitement par enzalutamide. Chez les patients devant recevoir un inhibiteur puissant du CYP2C8 de façon concomitante, la dose d'enzalutamide doit être réduite à 80 mg en une prise quotidienne (voir la rubrique Posologie et mode d'administration).
Inhibiteurs du CYP3A4
Le CYP3A4 joue un rôle mineur dans le métabolisme de l'enzalutamide. Après administration par voie orale d'itraconazole (200 mg en une prise quotidienne), un inhibiteur puissant du CYP3A4, chez des sujets sains de sexe masculin, l'ASC de l'enzalutamide a été augmentée de 41 %, tandis que sa Cmax est restée inchangée. L'ASC de la somme des fractions libres de l'enzalutamide et de son métabolite actif, a été augmentée de 27 % tandis que la Cmax est également restée inchangée. Aucune adaptation posologique n'est nécessaire lorsque Xtandi est administré en association avec des inhibiteurs du CYP3A4.
Inducteurs du CYP2C8 et du CYP3A4
Après administration par voie orale de rifampicine (600 mg en une prise quotidienne), un inducteur modéré du CYP2C8 et inducteur puissant du CYP3A4, chez des sujets sains de sexe masculin, l'ASC de l'enzalutamide et de son métabolite actif a diminué de 37 % tandis que la Cmax est restée inchangée. Aucune adaptation posologique n'est nécessaire lorsque Xtandi est administré en association avec des inducteurs du CYP2C8 ou du CYP3A4.
Effet potentiel de l'enzalutamide sur l'exposition d'autres médicaments
Induction enzymatique
L'enzalutamide est un inducteur enzymatique puissant qui augmente la synthèse de nombreux enzymes et transporteurs ; par conséquent, il faut s'attendre à des interactions avec des médicaments substrats des enzymes ou des transporteurs couramment utilisés. La diminution des concentrations plasmatiques peut être importante et annuler ou réduire l'effet clinique. Il existe également un risque de formation accrue de métabolites actifs. Les enzymes sur lesquelles l'enzalutamide est susceptible d'avoir un effet inducteur incluent : le CYP3A dans le foie et l'intestin, le CYP2B6, le CYP2C9, le CYP2C19 et l'uridine-5'- diphosphate glucuronosyltransférase (UGTs - enzyme de conjugaison des glucuronides). L'enzalutamide pourrait également avoir un effet inducteur sur la protéine de transport P-gp, de même que sur d'autres transporteurs tels que la protéine 2 associée à la multirésistance aux médicaments (MRP2), la protéine de résistance des cancers du sein (BCRP), et le polypeptide transporteur d'anions organiques 1B1 (OATP1B1).
Des études in vivo ont montré que l'enzalutamide est un inducteur puissant du CYP3A4 et un inducteur modéré du CYP2C9 et du CYP2C19. L'administration concomitante d'enzalutamide (160 mg en une prise quotidienne) et de doses orales uniques de substrats des CYP cibles chez des patients atteints d'un cancer de la prostate a induit une diminution de 86 % de l'ASC du midazolam (substrat du CYP3A4), de 56 % de l'ASC de la S-warfarine (substrat du CYP2C9) et de 70 % de l'ASC de l'oméprazole (substrat du CYP2C19). Il est possible que l'enzalutamide ait également un effet inducteur sur l'UGT1A1. Dans une étude clinique réalisée chez des patients atteints de cancer métastatique de la prostate résistant à la castration, Xtandi (160 mg une fois par jour) n'a pas eu d'effet cliniquement significatif sur la pharmacocinétique du docétaxel administré par voie intraveineuse (75 mg/m2 en perfusion toutes les 3 semaines). L'ASC du docétaxel a diminué de 12 % [Rapport des moyennes géométriques (RMG) = 0,882 (IC à 90 % : 0,767 ; 1,02)] tandis que la Cmax a baissé de 4 % [RMG = 0,963 (IC à 90 % : 0,834 ; 1,11)].
Il faut s'attendre à des interactions avec certains médicaments éliminés par métabolisme ou par transport actif. Il convient de ne pas utiliser ces médicaments ou de les utiliser avec prudence lorsque leurs effets thérapeutiques sur le patient sont importants et que leur posologie est difficilement ajustable sur la base du suivi de l'efficacité ou des concentrations plasmatiques. Certains éléments laissent penser que le risque d'atteinte hépatique après administration de paracétamol est plus élevé en cas d'administration concomitante d'inducteurs enzymatiques.
Les groupes de médicaments susceptibles d'être concernés incluent, entre autres (liste non limitative) :
des analgésiques (ex : fentanyl, tramadol)
des antibiotiques (ex : clarithromycine, doxycycline)
des agents anti-cancéreux (ex : cabazitaxel)
des antiépileptiques (ex : carbamazépine, clonazépam, phénytoïne, primidone, acide valproïque)
des antipsychotiques (ex : halopéridol)
des antithrombotiques (ex : acénocoumarol, warfarine, clopidogrel)
des bêtabloquants (ex : bisoprolol, propranolol)
des inhibiteurs calciques (ex : diltiazem, félodipine, nicardipine, nifédipine, vérapamil)
des glycosides cardiaques (ex : digoxine)
des corticoïdes (ex : dexaméthasone, prednisolone)
des traitements antirétroviraux contre le VIH (ex : indinavir, ritonavir)
des hypnotiques (ex : diazépam, midazolam, zolpidem)
un immunosuppresseur (ex : tacrolimus)
un inhibiteur de la pompe à protons (ex : oméprazole)
des statines métabolisées par le CYP3A4 (ex : atorvastatine, simvastatine)
des agents thyroïdiens (ex : lévothyroxine)
Il est possible que le potentiel d'induction enzymatique maximal de l'enzalutamide ne soit atteint qu'après un mois de traitement environ, lorsque les concentrations plasmatiques à l'état d'équilibre sont atteintes, bien que des effets inducteurs soient susceptibles d'apparaître plus tôt. Chez les patients prenant des médicaments substrats du CYP2B6, du CYP3A4, du CYP2C9, du CYP2C19 ou de l'UGT1A1, il faut évaluer la possible diminution des effets pharmacologiques (ou l'augmentation des effets en cas de formation de métabolites actifs) pendant le premier mois de traitement par enzalutamide et adapter la posologie si nécessaire. La demi- vie de l'enzalutamide étant longue (5,8 jours, voir la rubrique Propriétés pharmacocinétiques), il est possible que les effets sur les enzymes persistent pendant un mois ou plus après l'arrêt du traitement. Il pourrait s'avérer nécessaire de diminuer graduellement la dose du médicament concomitant à la fin du traitement par enzalutamide.
Substrats du CYP2C8 et du CYP1A2
L'enzalutamide (à la dose de 160 mg en une prise quotidienne) n'a pas provoqué de modification cliniquement significative de l'ASC ni de la Cmax de la caféine (substrat du CYP1A2) ou de la pioglitazone (substrat du CYP2C8). L'ASC de la pioglitazone a été augmentée de 20 %, tandis que la Cmax a diminué de 18 %. L'ASC et la Cmax de la caféine ont respectivement diminué de 11 % et 4 %. Aucune adaptation posologique n'est indiquée en cas d'administration concomitante de Xtandi et d'un substrat du CYP2C8 ou du CYP1A2.
Substrats de la P-gp
Les données in vitro indiquent que l'enzalutamide pourrait être un inhibiteur de la P-gp, un transporteur d'efflux. L'effet de l'enzalutamide sur les substrats de la P-gp n'a pas été étudié in vivo ; cependant, en clinique, l'enzalutamide pourrait être un inducteur de la P-gp par activation du récepteur nucléaire orphelin PXR (Pregnane X receptor). Les médicaments substrats de la P-gp ayant une marge thérapeutique étroite (ex : colchicine, étexilate de dabigatran, digoxine) doivent être utilisés avec prudence lorsqu'ils sont administrés avec Xtandi, et peuvent nécessiter une adaptation posologique pour maintenir les concentrations plasmatiques à un niveau optimal.
Substrats de la BCRP, de la MRP2, de l'OAT3 et de l'OCT1
Compte tenu des données in vitro, l'inhibition de la BCRP et de la MRP2 (dans l'intestin), ainsi que du transporteur d'anions organiques 3 (OAT3) et du transporteur de cations organiques 1 (OCT1) (au niveau systémique) ne peut être exclue. L'induction de ces transporteurs est également possible en théorie, et l'effet global est actuellement inconnu.
Médicaments allongeant l'intervalle QT
Le traitement par suppression androgénique étant susceptible d'allonger l'intervalle QT, l'utilisation concomitante de Xtandi avec des médicaments connus pour allonger l'intervalle QT, ou des médicaments capables d'induire des torsades de pointes tels que les antiarythmiques de classe IA (par exemple quinidine, disopyramide) ou de classe III (par exemple amiodarone, sotalol, dofetilide, ibutilide), la méthadone, la moxifloxacine, les antipsychotiques, etc. doit être évaluée avec précaution (voir rubrique Mises en garde et précautions d'emploi).
Effet de la nourriture sur l'exposition à l'enzalutamide
La nourriture n'a pas d'effet cliniquement significatif sur l'exposition à l'enzalutamide. Au cours des études cliniques, Xtandi a été administré sans tenir compte de la prise de nourriture.
Un traitement par enzalutamide doit être initié et supervisé par un médecin spécialiste expérimenté dans le traitement médical du cancer de la prostate.
Posologie
La dose recommandée est de 160 mg d'enzalutamide (quatre capsules molles de 40 mg) en une seule prise quotidienne par voie orale.
La castration médicale par un analogue de l'hormone de libération de la lutéinostimuline (LHRH) doit être maintenue pendant la durée du traitement pour les patients n'ayant pas subi de castration chirurgicale.
Si le patient oublie de prendre la dose prescrite de Xtandi à l'heure habituelle, elle doit être administrée aussi près que possible de l'heure habituelle de prise. Si le patient oublie de prendre la dose prescrite de Xtandi pendant toute une journée, il convient de reprendre le traitement le lendemain à la dose quotidienne habituelle.
En cas de toxicité de grade supérieur ou égal à 3 ou d'effet indésirable intolérable, il convient de suspendre le traitement pendant une semaine ou jusqu'à ce que les symptômes reviennent à un grade inférieur ou égal à 2, puis de reprendre le traitement à la même dose ou à une dose réduite si nécessaire (120 mg ou 80 mg).
Utilisation concomitante avec des inhibiteurs puissants du CYP2C8
L'utilisation concomitante d'inhibiteurs puissants du CYP2C8 doit être évitée autant que possible. Si le patient doit recevoir un inhibiteur puissant du CYP2C8 de façon concomitante, la dose d'enzalutamide doit être réduite à 80 mg en une prise quotidienne. En cas d'arrêt de l'administration concomitante de l'inhibiteur puissant du CYP2C8, l'enzalutamide doit être repris à la dose utilisée avant l'instauration de l'inhibiteur puissant du CYP2C8 (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Patients âgés
Aucune adaptation posologique n'est nécessaire chez les patients âgés (voir rubriques Propriétés pharmacodynamiques et Propriétés pharmacocinétiques).
Insuffisance hépatique
Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance hépatique légère, modérée ou sévère (respectivement classes A, B et C de Child-Pugh). Un allongement de la demi-vie de l'enzalutamide a toutefois été observé chez les patients présentant une insuffisance hépatique sévère (voir rubriques Mises en garde et précautions d'emploi et Propriétés pharmacocinétiques).
Insuffisance rénale
Aucune adaptation posologique n'est nécessaire chez les patients présentant une insuffisance rénale légère à modérée (voir rubrique Propriétés pharmacocinétiques). La prudence est recommandée chez les patients présentant une insuffisance rénale sévère ou de stade terminal (voir rubrique Mises en garde et précautions d'emploi).
Population pédiatrique
Il n'y a pas d'utilisation justifiée de l'enzalutamide dans la population pédiatrique dans l'indication du traitement du CPRC chez les hommes adultes.
Mode d'administration
Xtandi est à utiliser par voie orale. Les capsules molles ne doivent pas être mâchées, dissoutes ni ouvertes mais doivent être avalées entières avec de l'eau, et peuvent être prises avec ou sans nourriture.
Durée de conservation :
2 ans.
Précautions particulières de conservation :
Ce médicament ne nécessite pas de précautions particulières de conservation.
Sans objet.
Il n'existe pas d'antidote à l'enzalutamide. En cas de surdosage, l'administration d'enzalutamide doit être arrêtée et des mesures générales de prise en charge doivent être mises en place en tenant compte de la demi- vie de 5,8 jours. Après un surdosage, les patients peuvent être exposés à un risque accru de convulsions.
Classe pharmacothérapeutique: antihormones et apparentés, antiandrogènes, Code ATC: L02BB04
Mécanisme d'action
Le cancer de la prostate est un cancer sensible aux androgènes. Il répond à l'inhibition de la voie de signalisation des récepteurs aux androgènes. Malgré des taux sériques d'androgènes bas voire indétectables, la voie de signalisation des récepteurs aux androgènes continue de favoriser la progression de la maladie.
Pour stimuler la croissance des cellules tumorales, le récepteur aux androgènes doit pénétrer dans le noyau et se fixer à l'ADN. L'enzalutamide est un inhibiteur puissant de la voie de signalisation des récepteurs aux androgènes, qui en bloque plusieurs étapes. L'enzalutamide inhibe de façon compétitive la liaison des androgènes à leurs récepteurs, et inhibe donc la translocation nucléaire des récepteurs activés, et inhibe leur fixation à l'ADN et ce, même en cas de surexpression des récepteurs aux androgènes ou dans les cellules cancéreuses résistantes aux anti-androgènes. Le traitement par enzalutamide freine la croissance des cellules prostatiques cancéreuses et peut induire leur apoptose et la régression tumorale. Des études précliniques ont montré que l'enzalutamide n'a pas d'activité agoniste sur les récepteurs aux androgènes.
Effets pharmacodynamiques
Dans une étude clinique de phase III (AFFIRM) menée chez des patients après échec de la chimiothérapie à base de docétaxel, une diminution d'au moins 50 % du taux de PSA par rapport à la valeur initiale a été observée chez 54 % des patients recevant l'enzalutamide versus 1,5 % des patients recevant le placebo.
Dans une autre étude clinique de phase III (PREVAIL) menée chez des patients chimio-naïfs, les patients traités par enzalutamide présentaient un taux de réponse du PSA total significativement plus élevé (réponse définie par une réduction de ≥ 50 % par rapport à l'état initial) en comparaison des patients ayant reçu le placebo :78,0 % contre 3,5 % (différence = 74,5 %, p < 0,0001).
Dans une étude clinique de phase II (TERRAIN) menée chez des patients chimio-naïfs, les patients traités par enzalutamide présentaient un taux de réponse du PSA total significativement plus élevé (réponse définie par une réduction de ≥ 50 % par rapport à l'état initial) en comparaison des patients ayant reçu du bicalutamide : 82,1 % contre 20,9 % (différence = 61,2 %, p < 0,0001).
Dans une étude à un seul bras (9785-CL-0410) menée chez des patients préalablement traités pendant au moins 24 semaines par de l'abiratérone (plus prednisone), 22,4 % présentaient une diminution de ≥ 50 % du taux de PSA par rapport à l'état initial. Concernant les antécédents de chimiothérapie, la proportion de patients avec une diminution de ≥ 50 % du taux de PSA a été respectivement de 22,1 % et 23,2 % pour le groupe de patients sans antécédent de chimiothérapie et pour le groupe de patients avec un antécédent de chimiothérapie.
Dans l'étude clinique MDV3100-09 (STRIVE) dans le CPRC non métastatique et métastatique, les patients traités par enzalutamide présentaient un taux de réponse du PSA total confirmé significativement plus élevé (défini comme une réduction de ≥ 50 % par rapport à l'état initial), comparés aux patients traités par bicalutamide (81,3 % contre 31,3 % ; différence = 50,0 %, p < 0,0001).
Dans l'essai clinique MDV3100-14 (PROSPER) dans le CPRC non métastatique, les patients traités par enzalutamide présentaient un taux de réponse du PSA confirmé significativement plus élevé (défini comme une réduction de ≥ 50 % par rapport à l'état initial), comparés aux patients ayant reçu le placebo (76,3 % contre 2,4 % ; différence = 73,9 %, p < 0,0001).
Efficacité et sécurité cliniques
L'efficacité de l'enzalutamide a été établie au cours de trois études cliniques de phase III, multicentriques, randomisées, contrôlées versus placebo [MDV3100-14 (PROSPER), CRPC2 (AFFIRM), MDV3100-03 (PREVAIL)], chez des patients atteints d'un cancer de la prostate en progression après échec d'un traitement par suppression androgénique [analogue de la LHRH ou après orchidectomie bilatérale]. L'étude PREVAIL a été menée chez des patients atteints d'un CPRC métastatique n'ayant jamais reçu de chimiothérapie, tandis que l'étude AFFIRM a été menée chez des patients atteints d'un CPRC métastatique traités antérieurement par docétaxel et l'étude PROSPER a été menée chez des patients atteints d'un CPRC non métastatique. Tous les patients ont continué leur traitement par analogue de la LHRH ou avaient subi antérieurement une orchidectomie bilatérale. Dans le groupe du traitement actif, Xtandi a été administré par voie orale à une dose de 160 mg par jour. Dans les trois études cliniques, les patients du groupe témoin ont reçu un placebo. La prise de prednisone était autorisée, sans être obligatoire (dose maximale quotidienne autorisée : 10 mg de prednisone ou équivalent).
Les modifications du taux sérique de PSA pris isolément ne sont pas toujours prédictives du bénéfice clinique. Ainsi, dans ces trois études, il était recommandé que les patients poursuivent leurs traitements jusqu'à ce que les critères d'arrêt du traitement de l'étude soient remplis pour chaque étude comme spécifié ci-après.
Étude MDV3100-14 (PROSPER) (patients atteints d'un CPRC non métastatique)
L'étude PROSPER a été menée chez 1401 patients atteints d'un CPRC non métastatique à haut risque asymptomatique continuant un traitement par suppression androgénique (TSA ; défini comme un analogue de la LHRH ou une orchidectomie bilatérale antérieure). Les patients devaient avoir un temps de doublement du PSA ≤ 10 mois, un taux de PSA ≥ 2 ng/mL et une confirmation de maladie non métastatique par la revue centralisée indépendante en aveugle (blinded independent central review, BICR).
Les patients ayant des antécédents d'insuffisance cardiaque légère à modérée (classe NYHA 1 ou 2), et les patients prenant des médicaments connus pour abaisser le seuil épileptogène pouvaient être inclus. Les patients présentant des antécédents de convulsions ou une affection prédisposant aux convulsions et les patients ayant reçu certains traitements antérieurs pour le cancer de la prostate (à savoir, chimiothérapie, kétoconazole, acétate d'abiratérone, aminoglutéthimide et/ou enzalutamide) étaient exclus.
Les patients ont été randomisés selon un rapport de 2:1 pour recevoir soit l'enzalutamide à la dose de 160 mg en une prise orale quotidienne (N = 933), soit le placebo (N = 468). Les patients ont été stratifiés en fonction du temps de doublement de l'antigène spécifique de la prostate (Prostate Specific Antigen [PSA] Doubling Time [PSADT]) (< 6 mois ou ≥ 6 mois) et de l'utilisation d'agents ciblant l'os (oui ou non).
Les données démographiques et les caractéristiques de la maladie à l'état initial étaient réparties de façon équilibrée dans les deux bras de traitement. L'âge médian lors de la randomisation était de 74 ans dans le bras enzalutamide et de 73 ans dans le bras placebo. La plupart des patients (environ 71 %) de l'étude étaient d'origine ethnique caucasienne, 16 % d'origine ethnique asiatique et 2 % d'origine ethnique noire. Quatre- vingt-un pour cent (81 %) des patients avaient un score de performance ECOG de 0 et 19 % des patients avaient un score de performance ECOG de 1.
La survie sans métastase (metastasis-free survival, MFS) était le critère principal d'efficacité, définie comme le délai entre la randomisation et la progression radiologique ou le décès dans les 112 jours suivant l'arrêt du traitement sans preuve de progression radiologique, selon le premier événement survenu. Les critères secondaires d'efficacité clés évalués dans l'étude étaient le délai de progression du PSA, le délai d'instauration d'un nouveau traitement antinéoplasique (time to first use of new antineoplastic therapy, TTA), la survie globale (overall survival, OS). Les critères secondaires d'efficacité supplémentaires étaient le délai d'initiation d'une chimiothérapie cytotoxique et la survie sans chimiothérapie. Voir les résultats ci- dessous (tableau 2).
L'enzalutamide a montré une réduction statistiquement significative de 71 % du risque relatif de progression radiologique ou de décès versus placebo (HR = 0,29 (IC à 95 % =[0,24 ; 0,35]), p < 0,0001). La MFS médiane était de 36,6 mois (IC à 95 % =[33,1 ; non atteint]) dans le bras enzalutamide versus 14,7 mois (IC à 95 % =[14,2 ; 15,0]) dans le bras placebo. Des résultats cohérents pour la MFS ont également été observés dans tous les sous-groupes de patients prédéfinis sur la base du PSADT (< 6 mois ou ≥ 6 mois), de la région géographique (Amérique du Nord, Europe, reste du monde), de l'âge (< 75 ans ou ≥ 75 ans), de l'utilisation antérieure d'un agent ciblant l'os (oui ou non).
Tableau 2 : Résumé des résultats d'efficacité dans l'étude PROSPER (analyse sur la population en intention de traiter)
| | Enzalutamide N = 933 | Placebo N = 468 |
| Critère principal d'efficacité | ||
| Survie sans métastase | ||
| Nombre d'événements (%) | 219 (23,5) | 228 (48,7) |
| Médiane, mois (IC à 95 %)1 | 36,6 (33,1 ; NA) | 14,7 (14,2 ; 15,0) |
| Hazard ratio (IC à 95 %)2 | 0,29 (0,24 ; 0,35) | |
| Valeur de p3 | p < 0,0001 | |
| Critères secondaires d'efficacité clés | ||
| Délai de progression du PSA | ||
| Nombre d'événements (%) | 208 (22,3) | 324 (69,2) |
| Médiane, mois (IC à 95 %)1 | 37,2 (33,1 ; NA) | 3,9 (3,8 ; 4,0) |
| Hazard ratio (IC à 95 %)2 | 0,07 (0,05 ; 0,08) | |
| Valeur de p3 | p < 0,0001 | |
| Délai d'instauration d'un nouveau traitement antinéoplasique | ||
| Nombre d'événements (%) | 142 (15,2) | 226 (48,3) |
| Médiane, mois (IC à 95 %)1 | 39,6 (37,7 ; NA) | 17,7 (16,2 ; 19,7) |
| Hazard ratio (IC à 95 %)2 | 0,21 (0,17 ; 0,26) | |
| Valeur de p3 | p < 0,0001 | |
NA = non atteint.
Sur la base des estimations de Kaplan-Meier.
Le HR est basé sur un modèle de régression de Cox (avec le traitement comme seule covariable) stratifié en fonction du temps de doublement du PSA et de l'utilisation antérieure ou concomitante d'un agent ciblant l'os. Le HR est exprimé versus placebo, une valeur de < 1 étant en faveur de l'enzalutamide.
La valeur de p est dérivée d'un test de log-rank stratifié sur la base du temps de doublement du PSA (< 6 mois, ≥ 6 mois) et de l'utilisation antérieure ou concomitante d'un agent ciblant l'os (oui, non).
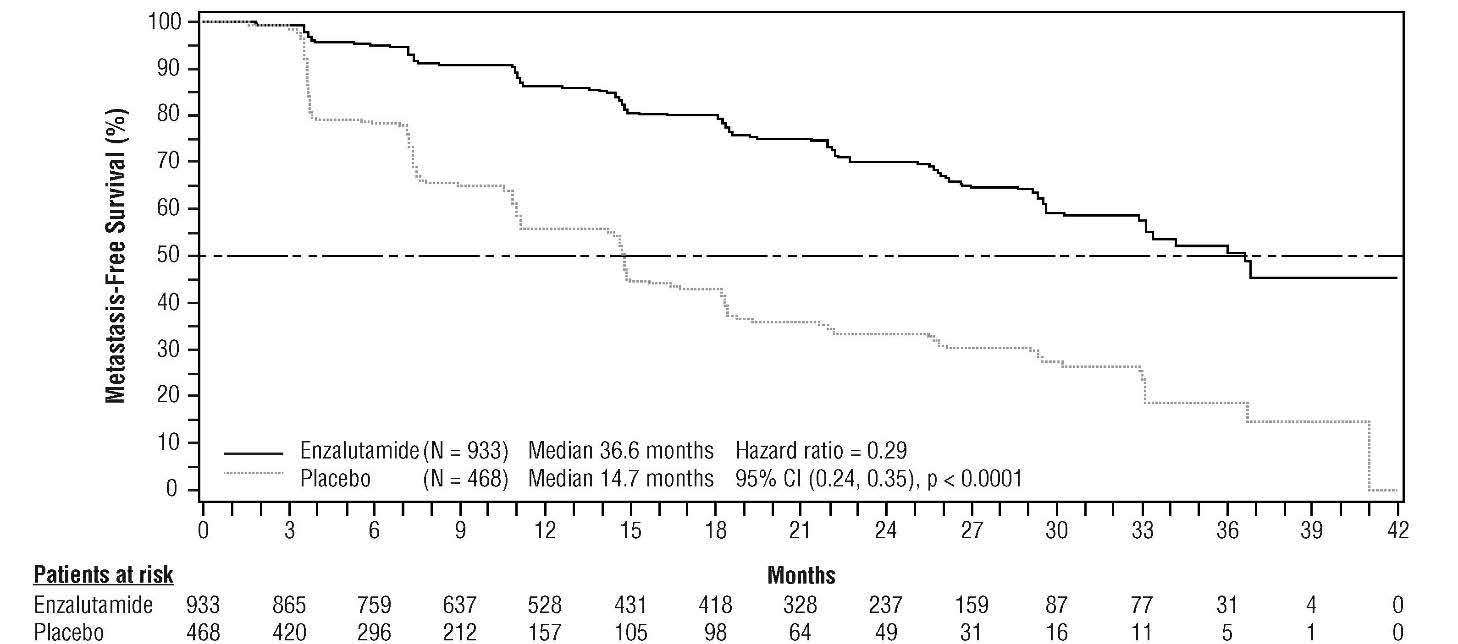
Figure 1 : Courbes de Kaplan-Meier sur la survie sans métastase dans l'étude PROSPER (analyse sur la population en intention de traiter)
Deux analyses intermédiaires prédéfinies nt été conduites à ce jour pour la survie globale ; la première au moment de l'analyse finale de la MFS (n = 165) (HR = 0,80 (IC à 95 % =[0,58 ; 1,09], p = 0,1519) et la seconde analyse intermédiaire (n = 288) (HR = 0,83 (IC à 95 % =[0,65 ; 1,06], p = 0,1344). La médiane n'a été atteinte dans aucun des deux groupes de traitement et aucune des deux analyses n'a révélé de différence statistiquement significative entre les groupes de traitement.
L'enzalutamide a montré une réduction statistiquement significative de 93 % du risque relatif de progression du PSA versus placebo (HR = 0,07 (IC à 95 % =[0,05 ; 0,08], p < 0,0001). Le délai médian jusqu'à la progression du PSA était de 37,2 mois (IC à 95 % =[33,1 ; non atteint]) dans le bras enzalutamide versus 3,9 mois (IC à 95 % =[3,8 ; 4,0]) dans le bras placebo.
L'enzalutamide a montré un allongement statistiquement significatif du délai d'instauration d'un nouveau traitement antinéoplasique versus placebo ([HR = 0,21 (IC à 95 % =[0,17 ; 0,26], p < 0,0001). Le délai médian d'instauration d'un nouveau traitement antinéoplasique était de 39,6 mois (IC à 95 % : 37,7 ; non atteint) dans le bras enzalutamide versus 17,7 mois (IC à 95 % =[16,2 ; 19,7]) dans le bras placebo.
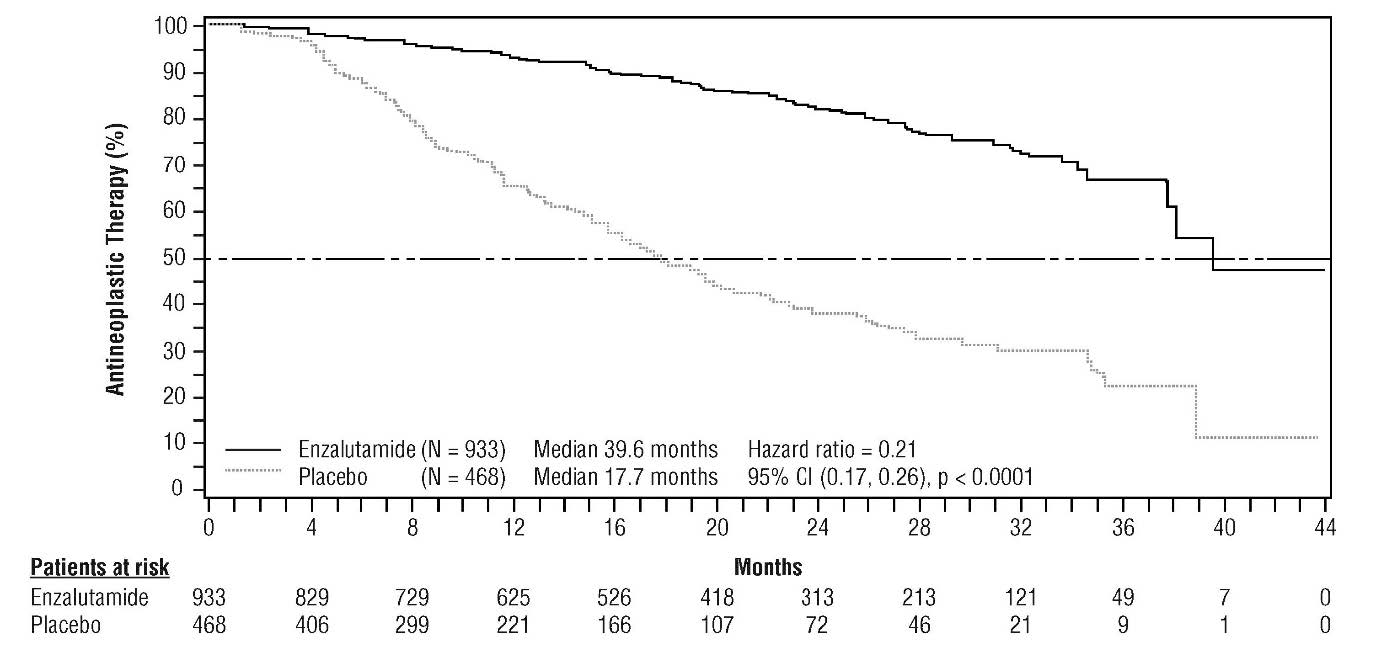
Figure 2 : Courbes de Kaplan-Meier sur le délai d'instauration d'un nouveau traitement antinéoplasique dans l'étude PROSPER (analyse sur la population en intention de traiter)
Étude MDV3100-09 (STRIVE) (patients n'ayant jamais reçu de chimiothérapie atteints d'un CPRC non métastatique/métastatique)
L'étude STRIVE a été menée chez 396 patients atteints d'un CPRC non métastatique ou métastatique présentant une progression biologique ou radiologique de la maladie malgré une première ligne de traitement par suppression androgénique qui ont été randomisés pour recevoir soit l'enzalutamide à la dose de 160 mg en une prise orale quotidienne (N = 198) soit le bicalutamide à la dose de 50 mg en une prise orale quotidienne (N = 198). La survie sans progression (PFS) était le critère d'évaluation principal, définie comme le délai entre la randomisation et la première preuve objective de progression radiologique, de progression du PSA ou de décès en cours d'étude. La PFS médiane était de 19,4 mois (IC à 95 % =[16,5 ; non atteint])) dans le groupe enzalutamide versus 5,7 mois (IC à 95 % =[5,6 ; 8,1]) dans le groupe bicalutamide (HR = 0,24 (IC à 95 % =[0,18 ; 0,32], p < 0,0001). Un bénéfice systématique de l'enzalutamide versus le bicalutamide sur la PFS a été observé dans tous les sous-groupes de patients prédéfinis. Concernant le sous-groupe non métastatique (N = 139), au total 19 des 70 (27,1 %) patients traités par enzalutamide et 49 des 69 (71,0 %) patients traités par bicalutamide ont présenté des événements de PFS (68 événements au total). Le hazard ratio était de 0,24 (IC à 95 % =[0,14 ; 0,42]) et le délai médian jusqu'à un événement de PFS n'a pas été atteint dans le groupe enzalutamide versus 8,6 mois dans le groupe bicalutamide.
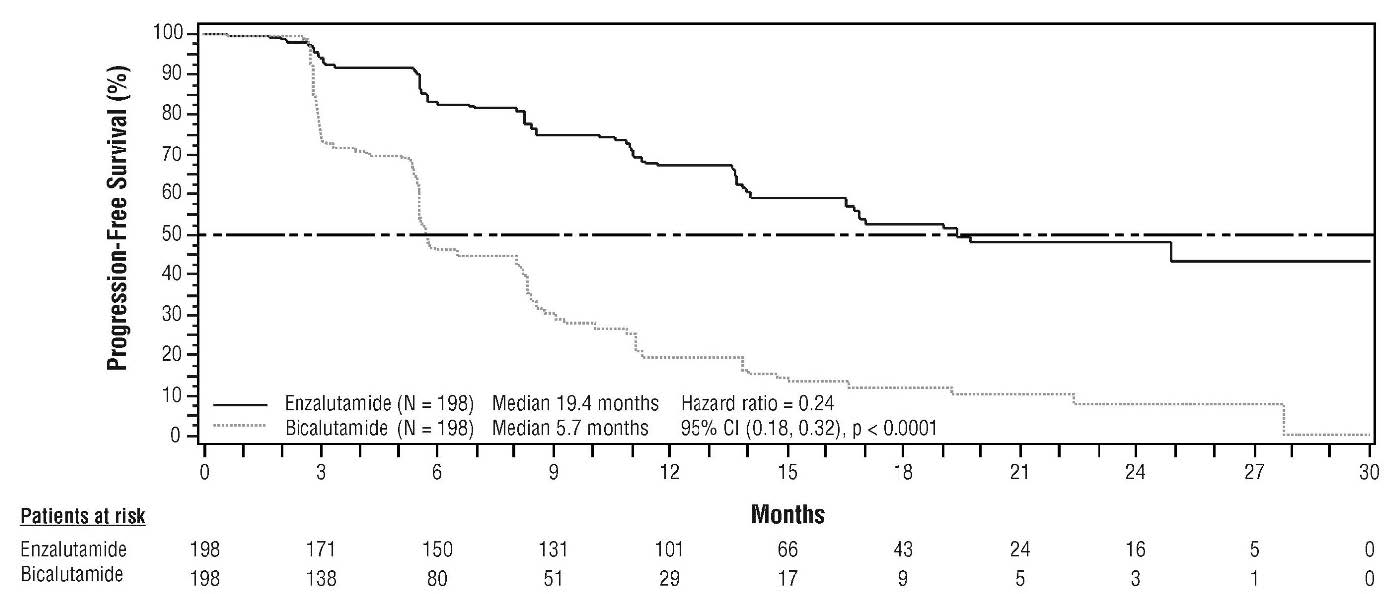
Figure 3 : Courbes de Kaplan-Meier sur la survie sans progression dans l'étude STRIVE (analyse sur la population en intention de traiter)
Étude 9785-CL-0222 (TERRAIN) (patients n'ayant jamais reçu de chimiothérapie atteints d'un CPRC métastatique)
L'étude TERRAIN a été menée chez 375 patients n'ayant jamais reçu de chimiothérapie et de traitement par anti-androgène atteints d'un CPRC métastatique qui ont été randomisés pour recevoir soit l'enzalutamide à la dose de 160 mg en une prise orale quotidienne (N = 184) soit le bicalutamide à la dose de 50 mg en une orale quotidienne (N = 191). La PFS médiane était de 15,7 mois pour les patients traités par enzalutamide versus 5,8 mois pour les patients traités par bicalutamide (HR = 0,44 (IC à 95 % =[0,34 ; 0,57], p < 0,0001). La survie sans progression a été définie comme la preuve objective d'une progression radiologique de la maladie par la revue centralisée indépendante, d'un événement osseux, de l'instauration d'un nouveau traitement antinéoplasique ou d'un décès de toute cause, selon le premier événement survenu. Un bénéfice systématique en termes de PFS a été observé dans tous les sous-groupes de patients prédéfinis.
Étude MDV3100-03 (PREVAIL) (patients atteints d'un CPRC métastatique n'ayant pas eu de chimiothérapie antérieure)
Un total de 1717 patients asymptomatiques ou peu symptomatiques n'ayant pas reçu de chimiothérapie antérieure ont été randomisés selon un rapport 1:1 pour recevoir soit enzalutamide, à une dose de 160 mg administrée par voie orale une fois par jour (N = 872), soit un placebo, pris une fois par jour par voie orale (N = 845). Les patients atteints de métastases viscérales, les patients ayant des antécédents d'insuffisance cardiaque légère à modérée (classe NYHA I ou II), et les patients prenant des médicaments connus pour abaisser le seuil épileptogène pouvaient être inclus. Les patients présentant des antécédents de convulsions ou une affection prédisposant aux convulsions et les patients souffrant de douleurs modérées ou sévères dues au cancer de la prostate étaient exclus. Le traitement à l'étude a été poursuivi jusqu'à la progression de la maladie (progression radiologique, événement osseux, ou progression clinique) et l'instauration d'une chimiothérapie cytotoxique ou d'un médicament expérimental, ou jusqu'à la survenue d'un événement indésirable justifiant l'arrêt du traitement.
Les données démographiques et les caractéristiques de la maladie à l'état initial étaient réparties de façon équilibrée dans les deux bras de traitement. L'âge médian était de 71 ans (valeurs extrêmes : 42 à 93 ans), 77 % des patients étant d'origine ethnique caucasienne, 10 % d'origine ethnique asiatique, 2 % d'origine ethnique noire, et 11 % d'autres groupes ethniques. Le score ECOG était de 0 chez soixante-huit pour cent (68 %) des patients et de 1 chez 32 % des patients. Le score du Questionnaire d'évaluation de la douleur (Brief Pain Inventory) était de 0-1 (asymptomatique) chez 67 % des patients et de 2-3 (peu symptomatique) chez 32 % des patients (douleur la plus intense rapportée par le patient au cours des 24 dernières heures, sur une échelle de 0 à 10). Environ 45 % des patients présentaient des lésions des tissus mous mesurables à l'entrée dans l'étude, et 12 % des patients présentaient des métastases viscérales (poumons et/ou foie).
Les co-critères principaux d'efficacité étaient la survie globale et la survie sans progression radiologique (rPFS). En plus de ces co-critères principaux, le bénéfice était également évalué par le délai d'initiation d'une chimiothérapie cytotoxique, la meilleure réponse globale au niveau des tissus mous, le délai de survenue du premier événement osseux, la réponse du PSA (≥ 50 % de baisse par rapport à l'état initial), le délai de progression du PSA, et le délai de détérioration de la qualité de vie d'après le questionnaire FACT- P.
La survie sans progression radiologique a été évaluée à l'aide d'études d'imagerie séquentielle comme défini par les critères PCWG2 (pour les lésions osseuses) et/ou par les critères RECIST (Response Evaluation Criteria In Solid Tumors) modifiés (pour les lésions des tissus mous). L'analyse de la rPFS était basée sur une évaluation centralisée de la progression radiologique.
Lors de l'analyse intermédiaire de la survie globale telle que prédéfinie au protocole après constat de 540 décès, le traitement par enzalutamide a démontré une amélioration statistiquement significative de la survie globale par rapport au placebo avec une réduction de 29,4 % du risque de décès (HR=0,706 IC 95% =[0,60 ; 0,84] ; p < 0,0001). Une mise à jour de l'analyse de la survie a été menée après constat de 784 décès. Les résultats de cette analyse sont en adéquation avec ceux de l'analyse intermédiaire (Tableau 3, Figure 4). En date de la mise à jour de l'analyse, 52 % des patients traités par enzalutamide et 81 % des patients sous placebo avaient reçu des traitements ultérieurs pour le CPRC métastatique susceptibles de prolonger la survie globale.
Tableau 3 : survie globale des patients ayant reçu l'enzalutamide ou le placebo dans l'étude PREVAIL (analyse sur la population en intention de traiter)
| | Enzalutamide (N = 872) | Placebo (N = 845) |
| Analyse intermédiaire prédéfinie | ||
| Nombre de décès (%) | 241 (27,6 %) | 299 (35,4 %) |
| Survie médiane, en mois (IC à 95 %) | 32,4 (30,1 ; NA) | 30,2 (28,0 ; NA) |
| Valeur de p1 | p < 0,0001 | |
| Hazard ratio (IC à 95 %)2 | 0,71 (0,60 ; 0,84) | |
| Analyse actualisée de la survie | ||
| Nombre de décès (%) | 368 (42,2 %) | 416 (49,2 %) |
| Survie médiane, en mois (IC à 95 %) | 35,3 (32,2 ; NA) | 31,3 (28,8 ; 34,2) |
| Valeur de p1 | p = 0,0002 | |
| Hazard ratio (IC à 95 %)2 | 0,77 (0,67 ; 0,88) | |
NA = non atteint.
La valeur de p est dérivée d'un test de log-rank non stratifié.
Le hazard ratio est dérivé d'un modèle à risques proportionnels non stratifié. Un hazard ratio < 1 est en faveur de l'enzalutamide.
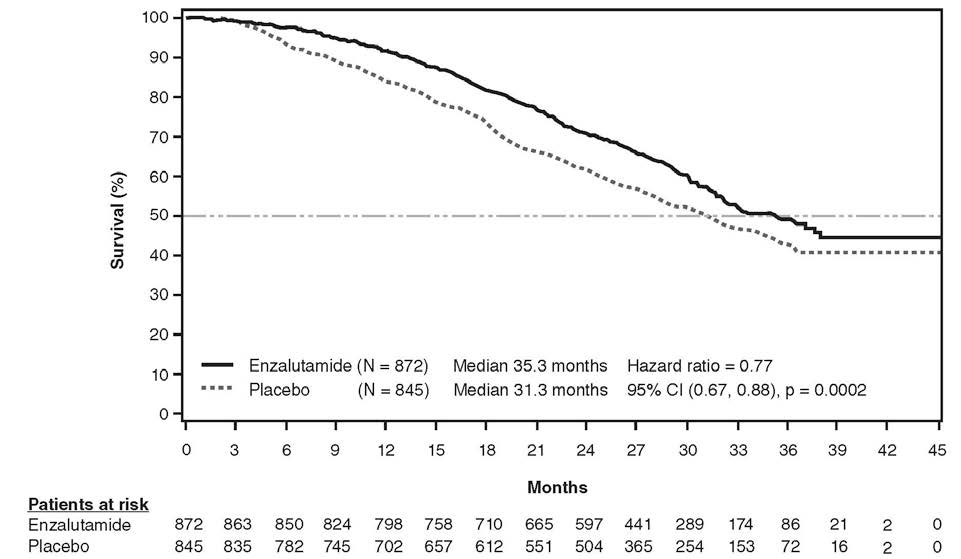
Figure 4 : Courbes de Kaplan-Meier sur la survie globale basées sur une analyse de survie actualisée dans l'étude PREVAIL (analyse sur la population en intention de traiter)
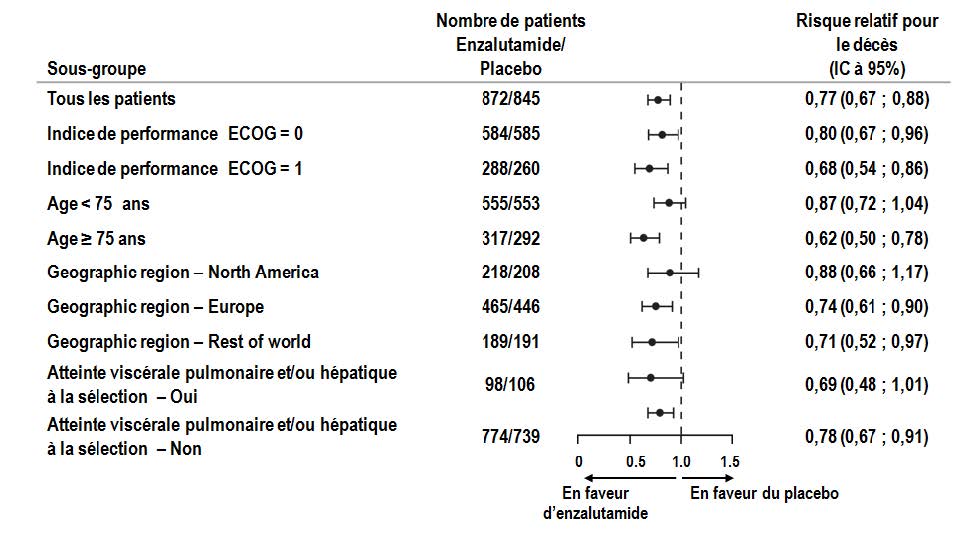
Figure 5 : Analyse de survie globale actualisée par sous-groupe : hazard ratio et intervalle de confiance à 95 % dans l'étude PREVAIL (analyse sur la population en intention de traiter)
Lors de l'analyse rPFS prédéfinie, une amélioration statistiquement significative a été démontrée entre les groupes de traitement avec une réduction de 81,4 % du risque de progression radiologique ou de décès [HR = 0,19 ; (IC 95 % = [0,15 ; 0,23]) ; p < 0,0001]. Cent dix-huit (14 %) des patients traités par enzalutamide et 321 (40 %) des patients sous placebo ont présenté un événement. La rPFS médiane n'a pas été atteinte (IC95 % = [13,8 ; non atteint]) dans le groupe traité par enzalutamide et était de 3,9 mois (IC 95 % = [3,7 ; 5,4]) dans le groupe sous placebo (figure 6). Un bénéfice homogène en termes de rPFS est observé dans tous les sous-groupes prédéfinis (par ex, âge, score ECOG initial, taux initiaux de PSA et LDH, score sur l'échelle de Gleason au diagnostic et atteinte viscérale à la sélection). Une analyse de suivi de la rPFS prévue au protocole et basée sur l'évaluation de la progression radiologique par les investigateurs démontre une amélioration statistiquement significative entre les groupes de traitement avec une réduction de 69,3 % du risque de progression radiologique ou de décès (HR = 0,31 ; (IC 95 % = [0,27 ; 0,35] ; p < 0,0001). La rPFS médiane était de 19,7 mois dans le groupe sous enzalutamide et de 5,4 mois dans le groupe sous placebo.
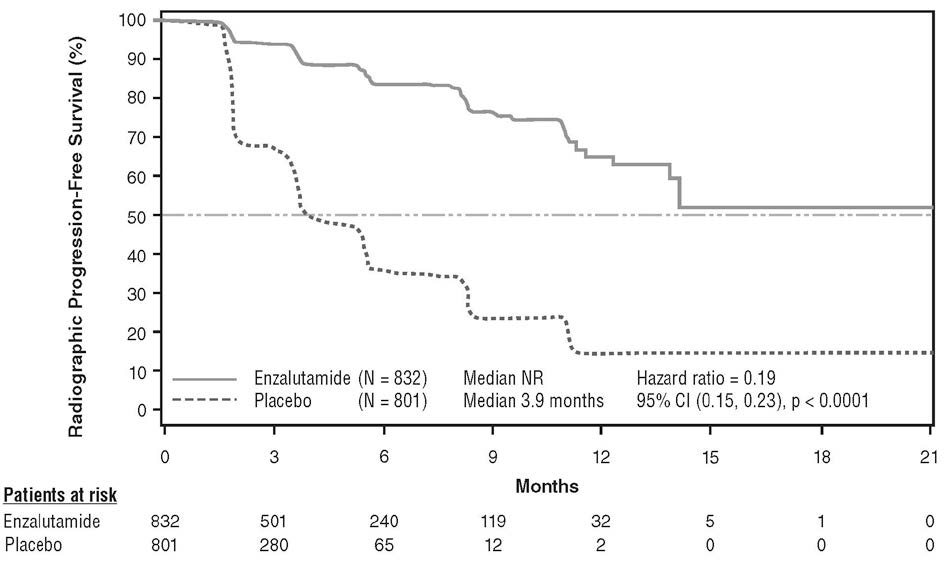
1633 patients étaient randomisés au moment de l'analyse principale de ce co-critère principal.
Figure 6 : Courbes de Kaplan-Meier sur la survie sans progression radiologique dans l'étude PREVAIL (analyse sur la population en intention de traiter)
Outre les co-critères principaux d'efficacité, des améliorations statistiquement significatives ont également été démontrées pour les critères suivants, déterminés de manière prospective.
La durée médiane jusqu'à l'initiation d'une chimiothérapie cytotoxique était de 28,0 mois pour les patients traités par enzalutamide et de 10,8 mois pour les patients sous placebo HR = 0,35 (IC 95 % = [0,30 ; 0,40]) ; p < 0,0001).
La proportion de patients traités par enzalutamide avec une maladie radiologiquement mesurable à l'état initial et qui présentaient une réponse objective au niveau des tissus mous était de 58,8 % (IC 95 % = [53,8 ; 63,7]) versus 5,0 % (IC 95 % = [3,0 ; 7,7]) pour les patients sous placebo. La différence absolue en termes de réponse objective au niveau des tissus mous entre les bras enzalutamide et placebo était de 53,9 % (IC 95 % = [48,5 ; 59,1], p < 0,0001). Des réponses complètes étaient rapportées chez 19,7 % des patients traités par enzalutamide versus 1,0 % des patients sous placebo, et des réponses partielles étaient rapportées chez39,1 % des patients traités par enzalutamide versus 3,9 % des patients sous placebo.
L'enzalutamide a induit une réduction significative de 28 % du risque de survenue du premier événement osseux (HR = 0,718 (IC 95 % = [0,61 ; 0,84] ; p < 0,0001). Un événement osseux était défini comme une radiothérapie ou une chirurgie osseuse du fait du cancer de la prostate, une fracture osseuse pathologique, une compression médullaire, ou une modification du traitement anticancéreux pour traiter la douleur osseuse. L'analyse incluait 587 événements osseux, parmi lesquels 389 (66,3 %) radiothérapies osseuses, 79 (13,5 %) compressions médullaires, 70 (11,9 %) fractures osseuses pathologiques, 45 (7,6 %) modifications du traitement anticancéreux pour traiter la douleur osseuse, et 22 (3,7 %) chirurgies osseuses.
Les patients traités par enzalutamide présentaient un taux de réponse du PSA total significativement plus élevé (défini comme une réduction de ≥ 50 % par rapport à l'état initial), comparés aux patients ayant reçu le placebo (78,0 % contre 3,5 % ; différence = 74,5 % ; p < 0,0001).
Le délai médian jusqu'à la progression du PSA selon les critères PCWG2 était de 11,2 mois pour les patients traités par enzalutamide et de 2,8 mois pour les patients ayant reçu le placebo (HR = 0,17 ; (IC 95 % =[0,15 ; 0,20] ; p < 0,0001).
Le traitement par enzalutamide a réduit le risque de dégradation du score FACT-P de 37,5 % par rapport au placebo (p < 0,0001). Le délai médian jusqu'à la dégradation du score FACT-P était de 11,3 mois dans le groupe enzalutamide et de 5,6 mois dans le groupe placebo.
Étude CRPC2 (AFFIRM) (patients atteints d'un CPRC métastatique ayant eu une chimiothérapie antérieure)
L'efficacité et la sécurité d'emploi de l'enzalutamide ont été évaluées dans le cadre d'une étude clinique de phase III, multicentrique, randomisée, contrôlée versus placebo, menée chez des patients atteints d'un CPRC métastatique ayant reçu du docétaxel et traités par un analogue de la LHRH ou ayant subi une orchidectomie. Au total, 1 199 patients ont été randomisés selon un rapport de 2:1 pour recevoir soit l'enzalutamide à la dose de 160 mg en une prise orale quotidienne (N = 800), soit le placebo en une prise quotidienne (N = 399). La prise de prednisone était autorisée, sans être obligatoire (dose maximale quotidienne autorisée : 10 mg de prednisone ou équivalent). Le protocole prévoyait que les patients des deux bras continuent le traitement jusqu'à la progression de la maladie (définie comme une progression radiologique confirmée ou la survenue d'un événement osseux) et l'instauration d'un nouveau traitement antinéoplasique systémique, jusqu'à l'apparition d'une toxicité inacceptable ou jusqu'à la sortie d'étude.
Les données démographiques et les caractéristiques de la maladie à l'état initial présentées ci-dessous étaient réparties de façon équilibrée dans les deux bras de traitement. L'âge médian était de 69 ans (valeurs extrêmes : 41 à 92 ans), 93 % des patients étant d'origine ethnique caucasienne, 4 % d'origine ethnique noire, 1 % d'origine ethnique asiatique et 2 % d'autres groupes ethniques. L'indice de performance ECOG était de 0 ou 1 chez 91,5 % des patients et de 2 chez 8,5 % des patients. Chez 28 % des patients, le score du Questionnaire d'évaluation de la douleur (Brief Pain Inventory) était supérieur ou égal à 4 (moyenne de la douleur la plus intense rapportée par le patient au cours des 24 dernières heures, calculée pendant 7 jours avant la randomisation). La plupart des patients (91 %) étaient atteints de métastases osseuses, et 23 % d'entre eux présentaient des métastases viscérales pulmonaires et/ou hépatiques. À l'entrée dans l'étude, 41 % des patients randomisés présentaient une progression du PSA uniquement, alors que 59 % présentaient une progression radiologique. Cinquante et un pour cent (51 %) des patients prenaient des bisphosphonates à l'état initial.
Les critères d'exclusion de l'étude AFFIRM comprenaient les affections prédisposant aux convulsions (voir rubrique Effets indésirables) et les médicaments connus pour abaisser le seuil épileptogène, ainsi que les maladies cardiovasculaires cliniquement significatives telles qu'une hypertension non contrôlée, des antécédents récents d'infarctus du myocarde ou d'angor instable, une insuffisance cardiaque de classe III ou IV selon les critères de la New York Heart Association (sauf si la fraction d'éjection était supérieure ou égale à 45 %), une arythmie ventriculaire cliniquement significative ou un bloc atrio-ventriculaire (sans pacemaker permanent).
Le protocole prévoyait la réalisation d'une analyse intermédiaire prédéfinie après 520 décès. Cette analyse a montré que la survie globale était supérieure chez les patients recevant l'enzalutamide par rapport aux patients sous placebo, de façon statistiquement significative (Tableau 4 et Figures 7 et 8).
Tableau 4 : Survie globale des patients ayant reçu l'enzalutamide ou le placebo dans l'étude AFFIRM (analyse sur la population en intention de traiter)
| | Enzalutamide (N = 800) | Placebo (N = 399) |
| Décès (%) | 308 (38,5 %) | 212 (53,1 %) |
| Survie médiane (mois) (IC à 95 %) | 18,4 (17,3 ; NA) | 13,6 (11,3 ; 15,8) |
| p1 | p < 0,0001 | |
| Hazard ratio (IC à 95 %)b | 0,63 (0,53 ; 0,75) | |
NA = non atteint.
La valeur de p est dérivée d'un test de log-rank stratifié selon l'indice de performance ECOG (0-1 vs 2) et le score de douleur moyen (< 4 versus ≥ 4).
Le hazard ratio est dérivé d'un modèle à risques proportionnels stratifié. Un hazard ratio < 1 est en faveur de l'enzalutamide.
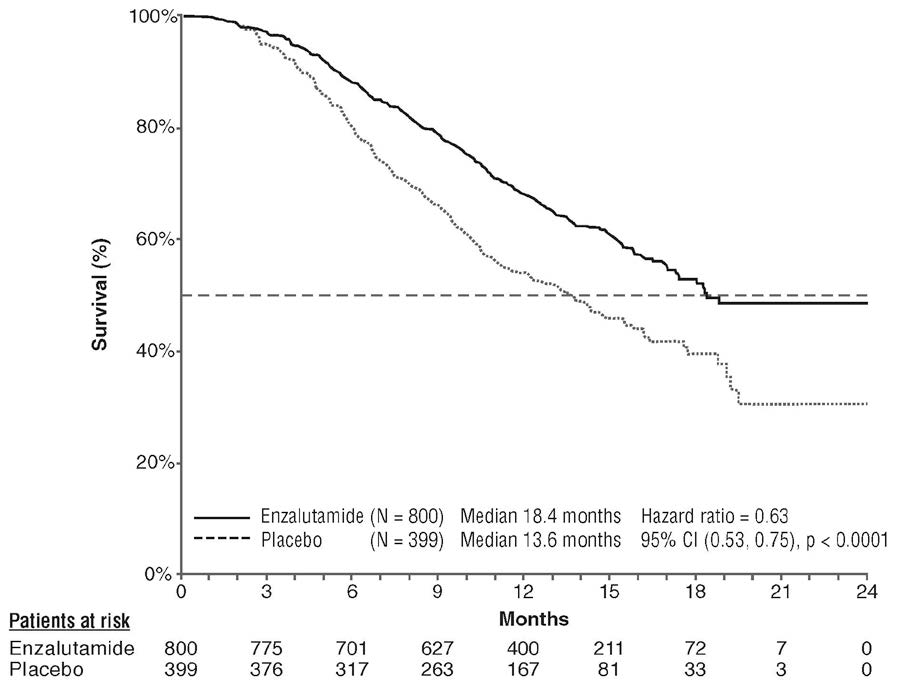
Figure 7 : Courbes de Kaplan-Meier sur la survie globale dans l'étude AFFIRM (analyse sur la population en intention de traiter)
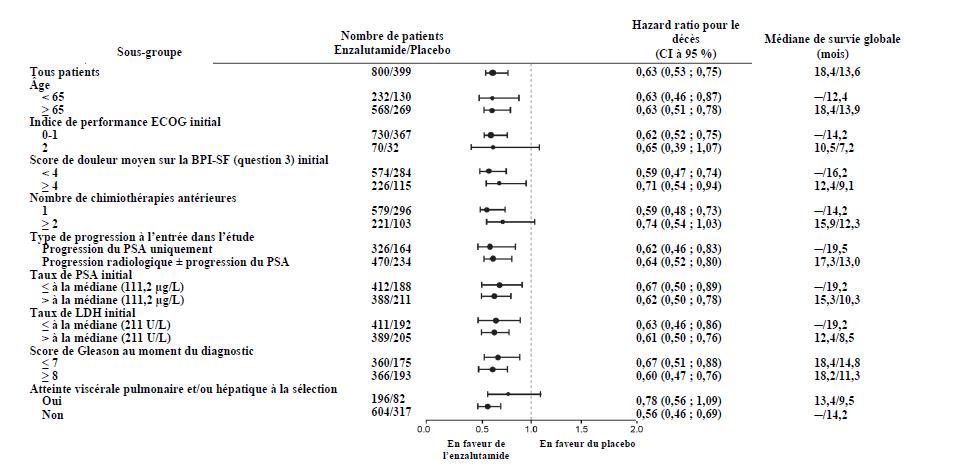
ECOG : Eastern Cooperative Oncology Group ; BPI-SF : version abrégée du questionnaire d'évaluation de la douleur (Brief Pain Inventory) ; PSA : antigène prostatique spécifique.
Figure 8 : Survie globale par sous-groupe dans l'étude AFFIRM : hazard ratio et intervalle de confiance à 95 %
Outre l'amélioration observée en matière de survie globale, les critères secondaires clés (progression du PSA, survie sans progression radiologique et délai d'apparition du premier événement osseux) étaient en faveur de l'enzalutamide, et étaient statistiquement significatifs après ajustement pour analyses multiples.
Après évaluation de la progression radiologique par l'investigateur au moyen des critères RECIST v.1.1 pour les tissus mous et en fonction de l'apparition d'au moins deux lésions osseuses sur la scintigraphie osseuse, la survie sans progression radiologique était de 8,3 mois pour les patients ayant reçu l'enzalutamide versus 2,9 mois pour les patients ayant reçu le placebo [HR = 0,40 (IC à 95 % =[0,35 ; 0,47]) ; p < 0,0001].
L'analyse comprenait 216 décès sans progression documentée et 645 événements de progression documentée parmi lesquels 303 (47 %) étaient dus à une progression des lésions des tissus mous, 268 (42 %) à une progression des lésions osseuses et 74 (11 %) aux deux.
La diminution confirmée des taux de PSA de 50 % et de 90 % était de 54,0 % et 24,8 % respectivement pour les patients traités par enzalutamide et de 1,5 % et 0,9 % respectivement pour les patients ayant reçu le placebo (p < 0,0001). Le délai médian jusqu'à la progression du PSA était de 8,3 mois pour les patients traités par enzalutamide et de 3,0 mois pour les patients ayant reçu le placebo (HR = 0,25 (IC à 95 %= [0,20 ; 0,30]) p < 0,0001).
Le délai médian jusqu'au premier événement osseux était de 16,7 mois pour les patients traités par enzalutamide et de 13,3 mois pour les patients ayant reçu le placebo [HR = 0,69 (IC à 95 % = [0,57 ; 0,84] p < 0,0001). Un événement osseux était défini comme une radiothérapie ou une chirurgie osseuse, une fracture osseuse pathologique, une compression médullaire ou la modification du traitement antinéoplasique pour traiter la douleur osseuse. L'analyse comprenait 448 événements osseux, parmi lesquels 277 (62 %) radiothérapies osseuses, 95 (21 %) compressions médullaires, 47 (10 %) fractures osseuses pathologiques, 36 (8 %) modifications du traitement antinéoplasique pour traiter la douleur osseuse et 7 (2 %) chirurgies osseuses.
Etude 9785-CL-0410 (enzalutamide post-abiratérone chez des patients atteints d'un CPRC métastatique)
Il s'agissait d'une étude à un seul bras chez 214 patients atteints d'un CPRC métastatique en progression traités par l'enzalutamide (160 mg une fois par jour) après au moins 24 semaines de traitement par l'acétate d'abiratérone plus prednisone. La médiane de rPFS (survie sans progression radiologique, critère principal d'évaluation de l'étude) a été de 8,1 mois (IC à 95 % =[6,1 ; 8,3]). La médiane de survie globale n'a pas été atteinte. La réponse du PSA (définie comme une diminution de ≥ 50 % par rapport à l'état initial) a été de 22,4 % (IC à 95 % =[17,0 ; 28,6]).
Pour les 69 patients préalablement traités par chimiothérapie, la médiane de rPFS a été de 7,9 mois (IC à 95 % =[5,5 ; 10,8]). La réponse du PSA a été de 23,2 % (IC à 95 % =[13,9 ; 34,9]).
Pour les 145 patients non préalablement traités par chimiothérapie, la médiane de rPFS a été de 8,1 mois (IC à 95 % =[5,7 ; 8,3]). La réponse du PSA a été de 22,1 % (IC à 95 % =[15,6 ; 29,7]).
Bien qu'il y ait une réponse limitée au traitement par enzalutamide après abiratérone chez certains patients, la raison de cette observation n'est actuellement pas connue. La méthodologie de l'étude n'a permis d'identifier ni les patients susceptibles de tirer bénéfice d'un traitement séquentiel par enzalutamide et abiratérone, ni l'ordre optimal de leur administration.
Patients âgés
Parmi les 3179 patients des essais cliniques contrôlés ayant reçu enzalutamide, 2518 (79 %) avaient 65 ans et plus, et 1162 (37 %) avaient 75 ans et plus. Aucune différence globale de sécurité et d'efficacité n'a été observée entre ces patients âgés et les patients plus jeunes.
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études réalisées avec l'enzalutamide dans tous les sous-groupes de la population pédiatrique dans le cancer de la prostate (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
L'enzalutamide est peu soluble dans l'eau. La solubilité de l'enzalutamide est augmentée par l'utilisation de macrogolglycérides caprylocapriques comme émulsifiants / agents tensioactifs. Au cours des études précliniques, l'enzalutamide a été mieux absorbé lorsqu'il était dissous dans des macrogolglycérides caprylocapriques.
La pharmacocinétique de l'enzalutamide a été évaluée chez des patients atteints d'un cancer de la prostate et chez des volontaires sains de sexe masculin. La demi-vie terminale moyenne (t1/2) de l'enzalutamide chez les patients après une dose unique par voie orale est de 5,8 jours (valeurs extrêmes : 2,8 à 10,2 jours) et l'état d'équilibre est atteint au bout d'un mois environ. En prise orale quotidienne, l'enzalutamide s'accumule selon un facteur de 8,3 environ par rapport à une dose unique. Les fluctuations des concentrations plasmatiques au cours de la journée sont faibles (indice de fluctuation de 1,25). La clairance de l'enzalutamide se fait principalement par métabolisme hépatique et produit un métabolite actif qui est aussi actif que l'enzalutamide et circule environ à la même concentration plasmatique.
Absorption
La concentration plasmatique maximale (Cmax) de l'enzalutamide est observée 1 à 2 heures après l'administration au patient. Dans l'étude d'équilibre de masse chez l'homme, l'absorption de l'enzalutamide administré par voie orale est estimée à 84,2 % au minimum. L'enzalutamide n'est pas un substrat des transporteurs d'efflux P-gp et BCRP. À l'état d'équilibre, les Cmax moyennes de l'enzalutamide et de son métabolite actif sont respectivement de 16,6 µg/mL (coefficient de variation [CV] : 23 %) et de 12,7 µg/mL (CV : 30 %).
La nourriture n'a aucun effet cliniquement significatif sur le degré d'absorption. Au cours des études cliniques, Xtandi a été administré sans tenir compte de la prise de nourriture.
Distribution
Chez les patients, le volume de distribution apparent moyen (VAD) de l'enzalutamide après administration orale d'une dose unique est de 110 L (CV : 29 %). Le volume de distribution de l'enzalutamide est supérieur au volume d'eau corporelle total, ce qui indique une distribution extravasculaire importante. Les études menées chez les rongeurs indiquent que l'enzalutamide et son métabolite actif peuvent traverser la barrière hémato-encéphalique.
L'enzalutamide est lié à hauteur de 97 % à 98 % aux protéines plasmatiques, principalement à l'albumine. Son métabolite actif est lié à 95 % aux protéines plasmatiques. In vitro, il n'y a pas eu de déplacement de la liaison protéique avec l'enzalutamide en présence d'autres médicaments à forte liaison protéique (warfarine, ibuprofène et acide salicylique) et réciproquement.
Biotransformation
L'enzalutamide est largement métabolisé. On retrouve deux métabolites majeurs dans le plasma humain : Le N-desmethyl enzalutamide (actif) et un dérivé de l'acide carboxylique (inactif). L'enzalutamide est métabolisé par le CYP2C8 et, dans une moindre mesure, par le CYP3A4/5 (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions) qui sont tous deux impliqués dans la formation du métabolite actif. In vitro, le N-desméthyl enzalutamide est métabolisé en acide carboxylique par la carboxylestérase 1, qui joue également un rôle mineur dans le métabolisme de l'enzalutamide en acide carboxylique. Le N-desmethyl enzalutamide n'était pas métabolisé par les CYP in vitro.
Dans les conditions d'une utilisation clinique, l'enzalutamide est un inducteur puissant du CYP3A4, un inducteur modéré du CYP2C9 et du CYP2C19 et n'a pas d'effet cliniquement significatif sur le CYP2C8 (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
Élimination
La clairance apparente moyenne (Cl/F) de l'enzalutamide chez les patients se situe entre 0,520 et 0,564 L/h.
Après administration orale d'enzalutamide marqué au 14C, 84,6 % de la radioactivité est retrouvée dans les 77 jours suivant l'administration : à 71,0 % dans les urines (principalement sous la forme du métabolite inactif, avec quelques traces d'enzalutamide et du métabolite actif) et à 13,6 % dans les fèces (0,39 % de la dose administrée sous forme inchangée).
Les données in vitro indiquent que l'enzalutamide n'est pas un substrat de l'OATP1B1, de l'OATP1B3 ni de l'OCT1, et que le N-desmethyl enzalutamide n'est pas un substrat des P-gp et BCRP.
Les données in vitro indiquent que l'enzalutamide et ses métabolites majeurs n'ont pas d'action inhibitrice sur les transporteurs suivants à des concentrations cliniquement significatives : OATP1B1, OATP1B3, OCT2 et OAT1.
Linéarité
Aucune déviation majeure de la proportionnalité à la dose n'a été observée dans la plage de doses comprise entre 40 et 160 mg. À l'état d'équilibre, les valeurs de Cmin de l'enzalutamide et de son métabolite actif chez chaque patient sont restées constantes pendant plus d'un an de traitement chronique, ce qui prouve la linéarité de la pharmacocinétique dans le temps, une fois l'état d'équilibre atteint.
Insuffisance rénale
Aucune étude n'a été réalisée avec l'enzalutamide chez des insuffisants rénaux. Les patients dont le taux de créatinine sérique était supérieur à 177 µmol/L (2 mg/dL) ont été exclus des études cliniques. Au vu d'une analyse pharmacocinétique de population, aucune adaptation posologique n'est nécessaire chez les patients dont la clairance de la créatinine calculée (ClCr) est supérieure ou égale à 30 mL/min (estimée au moyen de la formule de Cockcroft et Gault). L'enzalutamide n'a pas été évalué chez des patients atteints d'insuffisance rénale sévère (ClCr < 30 mL/min) ou d'une maladie rénale de stade terminal, et la prudence est recommandée lorsque ces patients sont traités. Il est peu probable que l'enzalutamide soit éliminé de façon significative lors d'une hémodialyse intermittente ou d'une dialyse péritonéale continue en ambulatoire.
Insuffisance hépatique
L'insuffisance hépatique n'a pas eu d'effet prononcé sur l'exposition totale à l'enzalutamide ou à son métabolite actif. La demi-vie de l'enzalutamide a toutefois été doublée chez les patients présentant une insuffisance hépatique sévère en comparaison avec les sujets sains du groupe témoin (10,4 jours versus 4,7 jours), en liaison peut-être avec une augmentation de la distribution tissulaire.
La pharmacocinétique de l'enzalutamide a été étudiée chez des patients présentant une insuffisance hépatique initiale légère (N = 6), modérée (N = 8) ou sévère (N=8) (respectivement classes A, B et C de Child-Pugh) et chez 22 sujets témoins appariés, présentant une fonction hépatique normale. Après administration orale d'une dose unique de 160 mg d'enzalutamide, l'ASC et la Cmax de l'enzalutamide ont été augmentées de 5 % et de 24 % respectivement chez les patients présentant une insuffisance hépatique légère, l'ASC a été augmentée de 29 % et la Cmax diminuée de 11 % chez les patients présentant une insuffisance hépatique modérée et l'ASC a été augmentée de 5 % et la Cmax diminuée de 41 % chez les patients présentant une insuffisance hépatique sévère, en comparaison avec les sujets sains du groupe témoin. Concernant la somme des fractions libres de l'enzalutamide et de son métabolite actif, l'ASC et la Cmax ont été augmentées de 14 % et 19 % respectivement chez les patients atteints d'une insuffisance légère, l'ASC a été augmentée de 14 % et la Cmax diminuée de 17 % chez les patients atteints d'une insuffisance modérée et l'ASC a été augmentée de 34 % et la Cmax diminuée de 27 % chez les patients atteints d'une insuffisance sévère, en comparaison avec les sujets sains du groupe témoin.
Origine ethnique
La majorité des patients inclus dans les études cliniques contrôlées (> 74 %) étaient de type caucasien. Sur la base de données pharmacocinétiques issues d'études menées chez des patients japonais et chinois atteints d'un cancer de la prostate, il n'y avait pas de différence cliniquement significative d'exposition au traitement entre les populations. Il n'y a pas suffisamment de données pour évaluer les différences potentielles dans la pharmacocinétique de l'enzalutamide chez les personnes d'autres origines ethniques.
Patients âgés
Aucun effet cliniquement significatif de l'âge sur la pharmacocinétique de l'enzalutamide n'a été observé lors de l'analyse de pharmacocinétique de population âgée.
Xtandi a une influence modérée sur l'aptitude à conduire des véhicules et à utiliser des machines, car des événements psychiatriques et neurologiques (notamment des convulsions) ont été rapportés (voir rubrique Effets indésirables). Les patients doivent être informés du risque potentiel de développer un événement psychiatrique ou neurologique pendant la conduite de véhicules ou l'utilisation de machines. Aucune étude n'a été menée pour évaluer les effets de l'enzalutamide sur l'aptitude à conduire des véhicules et à utiliser des machines.
Le traitement par enzalutamide de souris gravides a entraîné une augmentation de l'incidence de morts embryo-fœtales et des modifications externes et osseuses. Aucune étude de toxicité sur la reproduction n'a été réalisée avec l'enzalutamide. Cependant, lors des études menées chez le rat (4 et 26 semaines) et le chien (4, 13 et 39 semaines), des cas d'atrophie, d'aspermie/hypospermie et d'hypertrophie/hyperplasie du système reproducteur ont été observés, ce qui concorde avec l'activité pharmacologique de l'enzalutamide. Lors des études menées chez la souris (4 semaines), chez le rat (4 et 26 semaines) et chez le chien (4, 13 et 39 semaines), les modifications des organes reproducteurs associées à la prise d'enzalutamide ont été des diminutions de la masse des organes avec atrophie de la prostate et des épididymes. Une hypertrophie et/ou une hyperplasie des cellules de Leydig ont été observées chez la souris (4 semaines) et chez le chien (39 semaines). D'autres modifications ont également été observées au niveau des tissus reproducteurs, notamment une hypertrophie/hyperplasie de la glande pituitaire, une atrophie des vésicules séminales chez les rats, une hypospermie ainsi qu'une dégénérescence des tubes séminifères chez les chiens. Des différences liées au sexe ont été observées au niveau des glandes mammaires chez le rat (atrophie chez les mâles et hyperplasie lobulaire chez les femelles). Pour chaque espèce, les modifications des organes reproducteurs concordaient avec l'activité pharmacologique de l'enzalutamide et ont été réversibles ou partiellement résolues après une période de récupération de huit semaines. Aucune autre modification clinique ou histopathologique d'autres systèmes d'organes, y compris du foie, n'a été rapportée dans ces deux espèces.
Les études sur des rates gravides ont montré que l'enzalutamide et/ou ses métabolites sont transférés aux fœtus. Après administration orale d'enzalutamide radiomarqué au 14C chez des rates à 14 jours de gestation à la dose de 30 mg/kg (~ 1,9 fois la dose maximale indiquée chez l'humain), la radioactivité maximale dans le fœtus était atteinte 4 heures après l'administration et était inférieure à celle présente dans le plasma maternel avec un ratio tissu/plasma de 0,27. La radioactivité dans le fœtus diminuait à 0,08 fois la concentration maximale 72 heures après l'administration.
Les études sur des rates allaitantes ont montré que l'enzalutamide et/ou ses métabolites sont sécrétés dans le lait. Après administration orale d'enzalutamide radiomarqué au 14C à des rates allaitantes à la dose de 30 mg/kg (~ 1,9 fois la dose maximale indiquée chez l'humain), la radioactivité maximale dans le lait était atteinte 4 heures après l'administration et était 3,54 fois supérieure à celle présente dans le plasma maternel. Les résultats de l'étude ont également montré que l'enzalutamide et/ou ses métabolites sont transférés dans les tissus du raton via le lait et sont ensuite éliminés.
L'enzalutamide ne s'est pas montré génotoxique dans une batterie standard de tests in vitro et in vivo.Dans une étude de 6 mois menée chez des souris transgéniques rasH2, l'enzalutamide n'était pas cancérigène (absence d'observations néoplasiques) à des doses atteignant 20 mg/kg par jour (ASC24h ~317 µg.h/mL), qui se sont traduites par des niveaux d'exposition plasmatique similaires à l'exposition clinique (ASC24h 322 µg.h/mL) chez les patients atteints de CPRCm traités par une dose de 160 mg en une prise orale quotidienne. Le traitement quotidien de rats pendant deux ans par l'enzalutamide à la dose de 10- 100 mg/kg/jour a entraîné une incidence accrue de plusieurs tumeurs principalement bénignes. Les plus importantes d'entre elles ont été des tumeurs bénignes des cellules de Leydig, un papillome urothélial et un carcinome de la vessie. Les tumeurs bénignes des cellules de Leydig sont prévisibles d'après les propriétés pharmacologiques de cet anti-androgène et ne sont pas considérées comme pertinentes pour l'être humain. Des papillomes urothéliaux et carcinomes de la vessie sont attendus chez les rats en raison de la structure horizontale de la vessie du rat, pouvant présenter une urine concentrée et une irritation prolongée provoquées par les calculs. Dans l'étude, des calculs et des cristaux ont été observés dans la vessie des rats. Toutefois, aucun rationnel mécanistique évident expliquant spécifiquement cette malignité ne peut être établi, et compte tenu du fait que les niveaux d'exposition à l'enzalutamide et à ses métabolites, d'après l'ASC obtenue au cours de l'étude, étaient inférieurs ou similaires à ceux des patients atteints d'un cancer de la prostate à la dose recommandée de 160 mg/jour, on ne peut exclure la possibilité que l'enzalutamide ait des effets cancérogènes sur la vessie humaine. Les autres tumeurs, qui sont aussi potentiellement liées à la pharmacologie primaire de l'enzalutamide, sont le fibroadénome mammaire et le thymome bénin chez les mâles, les tumeurs bénignes de l'ovaire à cellules de la granulosa chez les femelles et l'adénome hypophysaire de la pars distalis chez les deux sexes. Les niveaux d'exposition atteints dans cette étude chez les rats mâles à la semaine 26 à la dose de 100 mg/kg/jour pour l'enzalutamide plus ses métabolites actifs M1 et M2 (ASC24 : enzalutamide ~457 µg.h/mL, M1 ~321 µg.h/mL, M2 ~35 µg.h/mL) étaient inférieurs ou égaux à ceux observés chez les patients atteints d'un cancer de la prostate traités à la dose recommandée (160 mg/jour) d'enzalutamide (ASC24 : enzalutamide ~322 µg.h/mL, M1 ~193 µg.h/mL, M2 ~278 µg.h/mL). L'enzalutamide n'était pas phototoxique lors des études in vitro.
Xtandi ne doit pas être manipulé par des personnes autres que le patient et ses aidants et, en particulier, par les femmes qui sont enceintes ou en âge de procréer. Les capsules molles ne doivent pas être dissoutes ou ouvertes.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Prescription initiale hospitalière annuelle, réservée aux spécialistes en oncologie ou aux médecins compétents en cancérologie.
Capsules molles, de forme oblongue (environ 20 mm x 9 mm), de couleur blanche à blanc cassé, avec la mention « ENZ » imprimée à l'encre noire sur une face.
Pochette en carton contenant 28 capsules molles sous plaquettes thermoformées (PVC/PCTFE/aluminium). Chaque boîte contient 4 pochettes (112 capsules molles).