

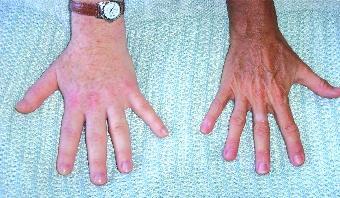

Circonstances de diagnostic
Le diagnostic est évoqué devant un syndrome dysmorphique marqué par, au niveau de l'extrémité céphalique, une implantation basse des cheveux, une hypertrophie des sinus frontaux, une saillie des arcades sourcilières, un nez épaté avec une hypertrophie des sillons naso-géniens, des lèvres épaisses et tombantes, un prognathisme avec une perte de l'articulé dentaire et une macroglossie. La comparaison à des photographies antérieures est souvent utile au diagnostic.
Au niveau des extrémités des membres par une infiltration des mains et des pieds avec augmentation de la taille des bagues et de la pointure des chaussures, des doigts en baguette de tambour, des mains en battoir, un épaississement du coussinet talonnier. Une cyphose ou une scoliose avec une projection en avant du sternum.
Le diagnostic d'acromégalie peut être fait devant des complications:
– cardio-vasculaires avec une cardiomyopathie fréquente et précoce chez les sujets jeunes, une hypertension artérielle présente chez 25 à 35 % des patients, une hypertrophie ventriculaire gauche, des troubles du rythme cardiaque, une insuffisance cardiaque réversible après contrôle de l'hypersécrétion somatotrope ;
– pulmonaires avec un syndrome d'apnée du sommeil, présent chez plus de 50 % des patients, responsable de troubles du sommeil, de ronflements, de céphalées matinales, ou plus rarement une insuffisance respiratoire ;
– rhumatologiques avec des atteintes périphériques ou rachidiennes responsables de douleurs articulaires ou de compressions nerveuses (syndrome du canal carpien, sciatalgie, névralgie cervico-brachiale) altérant la qualité de vie de ces patients ;
– digestives avec une fréquence accrue de polypes coliques, multiples, dysplasiques et de cancers coliques ;
– tumorales hypophysaires aiguës au cours d'une apoplexie hypophysaire ou chronique avec des céphalées, des troubles du champ visuel ou exceptionnellement des paralysies oculomotrices lors d'envahissement du sinus caverneux.
Ailleurs, le diagnostic doit être évoqué devant:
– une asthénie, parfois associée à un syndrome dépressif ;
– une viscéromégalie avec, outre la macroglossie, un goitre euthyroïdien, une hépatomégalie, un dolichomégacôlon ;
– des signes cutanés avec une peau épaisse, des sillons profonds, un cutis verticis gyrata du cuir chevelu, une hypertrichose ou un hirsutisme, un acanthosis nigricans, des fibromes sessiles ou pédiculés ;
– une hypersudation avec une odeur alliacée, une hypersécrétion séborrhée ;
– des signes gonadiques avec des troubles du cycle, un syndrome aménorrhée galactorrhée (normo ou hyperprolactinique), une diminution de la libido ou une impuissance ;
– une insuffisance anté-hypophysaire ;
– un giganto-acromégalisme chez un sujet prépubertaire.
Signes biologiques évocateurs
Enfin, le diagnostic d'acromégalie peut être évoqué devant des signes biologiques comme :
– une intolérance aux hydrates de carbone présente chez 60 % des patients ou un diabète sucré chez 13 à 32 % des sujets ;
– une hyperphosphorémie avec une hypercalciurie responsable de lithiases rénales ;
– une hypokaliémie avec une diminution de la rénine chez un patient hypertendu.
Diagnostic positif
Le diagnostic d'acromégalie doit être affirmé par les explorations paracliniques avec :
– une augmentation de la concentration de la GH (dosée avec une trousse calibrée par rapport au standard international 98/574) en situation basale et non freinée au cours de l'hyperglycémie provoquée par voie orale (GH < 1 µg/l ou < 0,3 µg/l selon les trousses utilisées) ;
– une augmentation de la concentration de l'IGF-1, marqueur d'activité périphérique de la GH, dont la concentration plasmatique doit être interprétée en fonction des techniques de dosage, de l'âge et du sexe des patients.
C'est ainsi que :
– une GH < 0,3 µg/l avec une IGF-1 normale (pour l'âge et le sexe du patient) élimine le diagnostic d'acromégalie,
– une GH élevée (> 2 µg/l) et non freinée par l'HGPO (> 0,3 µg/l) avec une IGF-1 élevée fait le diagnostic d'acromégalie ;
Après une chirurgie hypophysaire, le diagnostic d'acromégalie est fait sur l'examen immuno-histochimique d'un adénome hypophysaire mettant en évidence une fixation de l'anticorps anti-GH sur la majorité des cellules adénomateuses.
Diagnostic différentiel
Sur le plan clinique, l'acromégalie doit être différenciée des syndromes pseudo-acromégaliques généralisés ou localisés, constitutionnels ou acquis, au cours desquels l'exploration somatotrope est normale.
Une augmentation de la concentration plasmatique de la GH peut être observée au cours de la grossesse (interférence de dosage avec la GH placentaire), d'un traitement estrogénique, d'un stress, de la puberté, d'une insuffisance rénale chronique, d'une insuffisance cardiaque, d'un diabète décompensé, d'une malnutrition ou d'une anorexie.
La concentration plasmatique de l'IGF-1 est augmentée au cours du 2e trimestre de la grossesse (sous l'effet de la GH placentaire), de la puberté et d'une hyperthyroïdie.
Diagnostic étiologique
Le diagnostic d'acromégalie impose la réalisation d'une IRM de la région hypothalamo-hypophysaire qui pourra mettre en évidence :
– un adénome hypophysaire chez plus de 95 % des patients : de moins de 10 mm de diamètre ou micro-adénome chez 10 à 20 % des patients, de plus de 10 mm ou macro-adénome chez 75 à 80 % des patients acromégales. L'IRM précisera ainsi la taille de l'adénome, sa situation au sein de l'hypophyse ou éventuellement son caractère ectopique, l'extension supra-sellaire avec le risque de compression chiasmatique (qui sera objectivée par un examen du champ visuel), ou son invasion dans le sinus caverneux.
Les adénomes somatotropes peuvent être purs ou pluri-sécrétants (le plus souvent GH et prolactine), parfois responsables d'une insuffisance anté-hypophysaire. Ils sont le plus souvent sporadiques et rarement familiaux au cours des polyendocrinopathies de type Men 1, de la maladie de McCune Albright, du syndrome de Carney, ou dans le cadre des rares familles d'adénomes somatotropes (mutations du gène AIP) ;
– rarement un carcinome somatotrope lorsqu'il existe des métastases cérébrales ou extra-crâniennes, ou une hyperplasie hypophysaire liée à une hyperplasie des cellules somatotropes lors d'une sécrétion de GHRH par un gangliocytome hypothalamique, un carcinome bronchique, une tumeur endocrine du pancréas, un carcinome médullaire de la thyroïde, un phéochromocytome. Chez ces patients, le bilan devra comporter un dosage plasmatique du GHRH et un examen tomodensitométrique cervico-thoraco-abdominal parfois orienté par une scintigraphie à l'octréotide marqué.
Enfin, lorsque le bilan neuro-radiologique est normal, exceptionnellement une sécrétion extra-hypophysaire de GH peut être mise en évidence au cours de lymphomes ou de tumeurs pancréatiques.
Conclusion
L'acromégalie est une affection endocrinienne rare dont le diagnostic est souvent tardif au stade des complications.
Le diagnostic peut être fait par le médecin généraliste, mais également par de nombreux spécialistes (cardiologue, pneumologue, ORL, rhumatologue, dermatologue, ophtalmologue, gynécologue et endocrinologue) du fait du polymorphisme des signes cliniques et des complications liés à l'hypersécrétion somatotrope.
Il doit être évoqué sur l'association de signes tels que céphalées, hypersudation, syndrome du canal carpien bilatéral, asthénie. Il doit être précoce pour limiter la morbidité quand les lésions ne sont pas trop évoluées et donc réversibles.
Le diagnostic précoce d'acromégalie devrait augmenter la proportion des micro-adénomes hypophysaires dont le traitement de première intention est l'adénomectomie par voie rhinoseptale. Dans tous les autres cas, le contrôle de l'hypersécrétion somatotrope par le traitement chirurgical et médical (analogues de la somatostatine, agoniste dopaminergique, antagoniste du récepteur de la GH) améliorera la survie des patients acromégales.
Pause exceptionnelle de votre newsletter
En cuisine avec le Dr Dominique Dupagne
[VIDÉO] Recette d'été : la chakchouka
Florie Sullerot, présidente de l’Isnar-IMG : « Il y a encore beaucoup de zones de flou dans cette maquette de médecine générale »
Covid : un autre virus et la génétique pourraient expliquer des différences immunitaires, selon une étude publiée dans Nature